

The critical role of QM/MM X-ray refinement and accurate tautomer/protomer determination in structure-based drug design
- PMID: 33108589
- PMCID: PMC8018927
- DOI: 10.1007/s10822-020-00354-6
The critical role of QM/MM X-ray refinement and accurate tautomer/protomer determination in structure-based drug design
Abstract
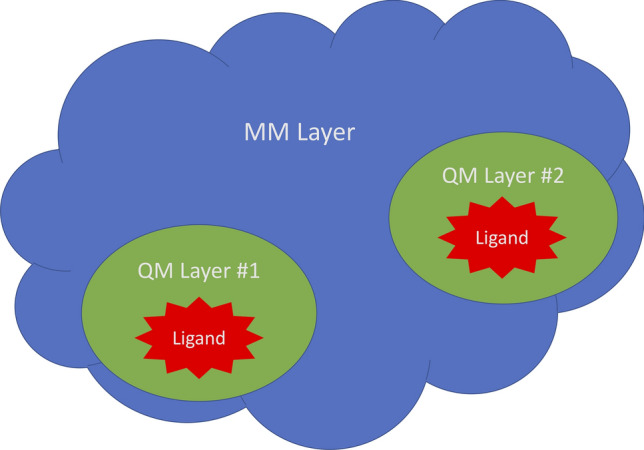
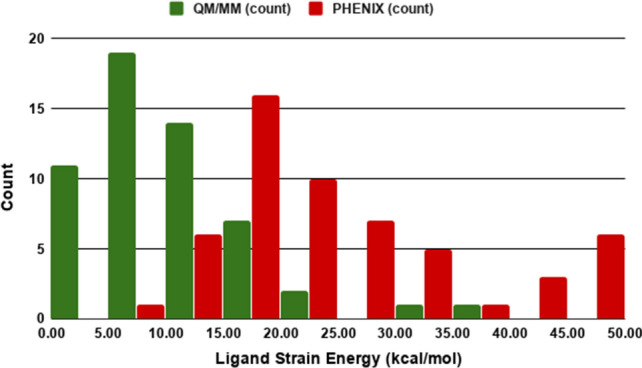
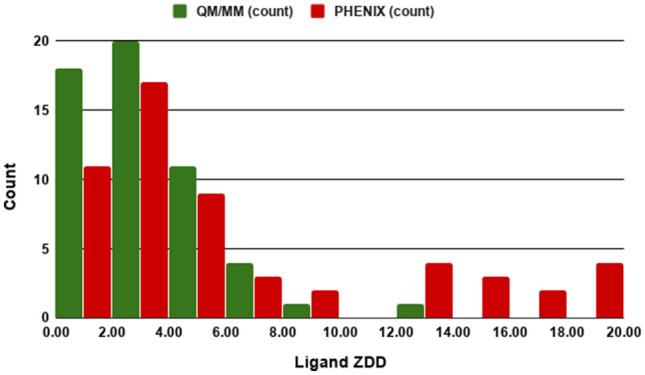
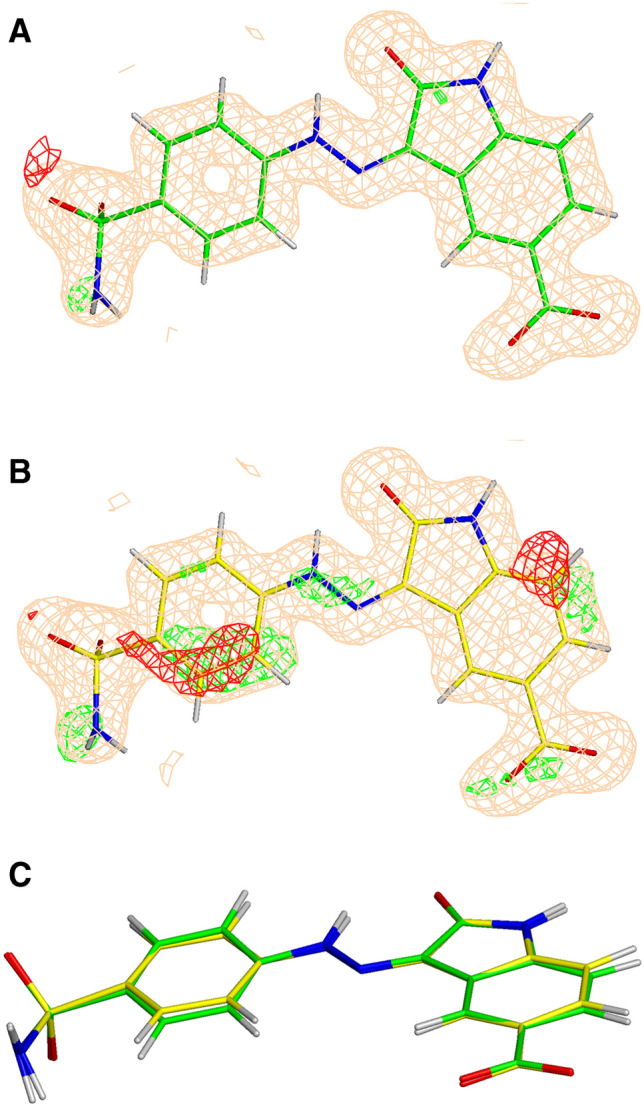
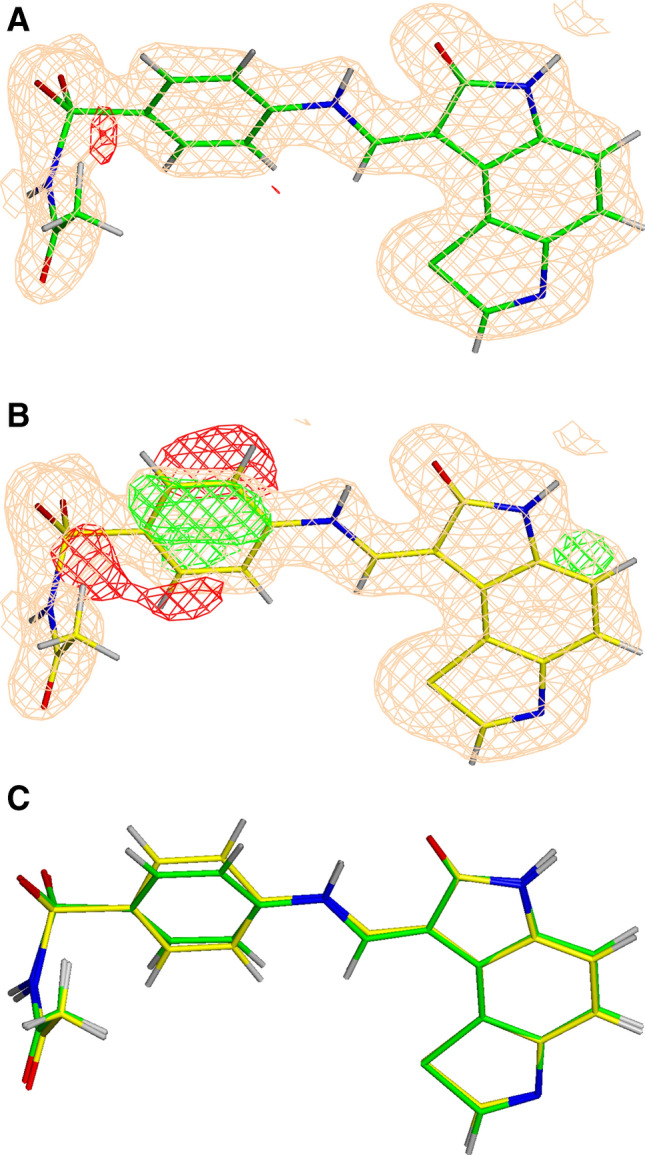
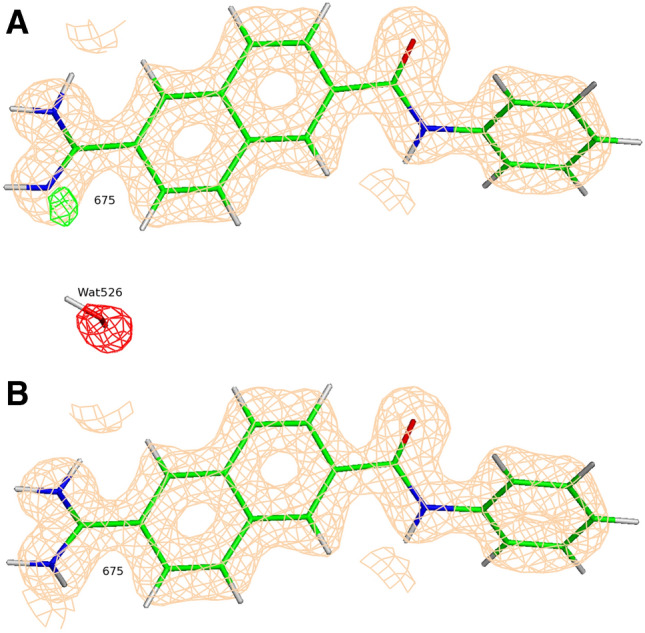
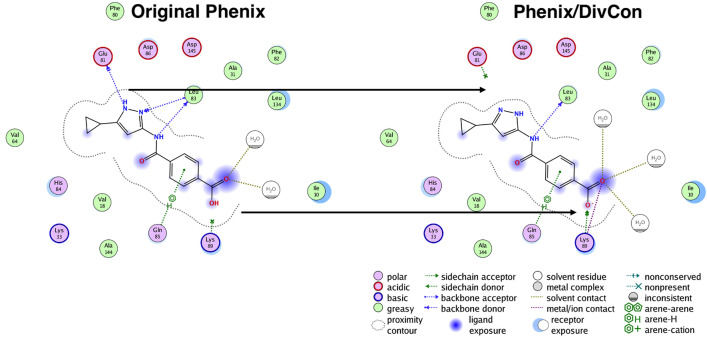
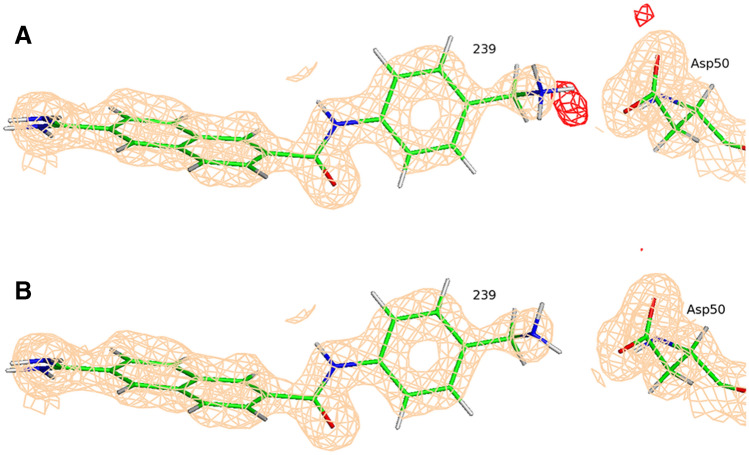
Conventional protein:ligand crystallographic refinement uses stereochemistry restraints coupled with a rudimentary energy functional to ensure the correct geometry of the model of the macromolecule-along with any bound ligand(s)-within the context of the experimental, X-ray density. These methods generally lack explicit terms for electrostatics, polarization, dispersion, hydrogen bonds, and other key interactions, and instead they use pre-determined parameters (e.g. bond lengths, angles, and torsions) to drive structural refinement. In order to address this deficiency and obtain a more complete and ultimately more accurate structure, we have developed an automated approach for macromolecular refinement based on a two layer, QM/MM (ONIOM) scheme as implemented within our DivCon Discovery Suite and "plugged in" to two mainstream crystallographic packages: PHENIX and BUSTER. This implementation is able to use one or more region layer(s), which is(are) characterized using linear-scaling, semi-empirical quantum mechanics, followed by a system layer which includes the balance of the model and which is described using a molecular mechanics functional. In this work, we applied our Phenix/DivCon refinement method-coupled with our XModeScore method for experimental tautomer/protomer state determination-to the characterization of structure sets relevant to structure-based drug design (SBDD). We then use these newly refined structures to show the impact of QM/MM X-ray refined structure on our understanding of function by exploring the influence of these improved structures on protein:ligand binding affinity prediction (and we likewise show how we use post-refinement scoring outliers to inform subsequent X-ray crystallographic efforts). Through this endeavor, we demonstrate a computational chemistry ↔ structural biology (X-ray crystallography) "feedback loop" which has utility in industrial and academic pharmaceutical research as well as other allied fields.
Keywords: CSAR set; Ligand strain; Quantum mechanics X-ray refinement; Structure guided drug discovery; Structure-based drug discovery; X-ray crystallography; difference density Z-score; high throughput crystallography; protonation states; tautomers.
Figures
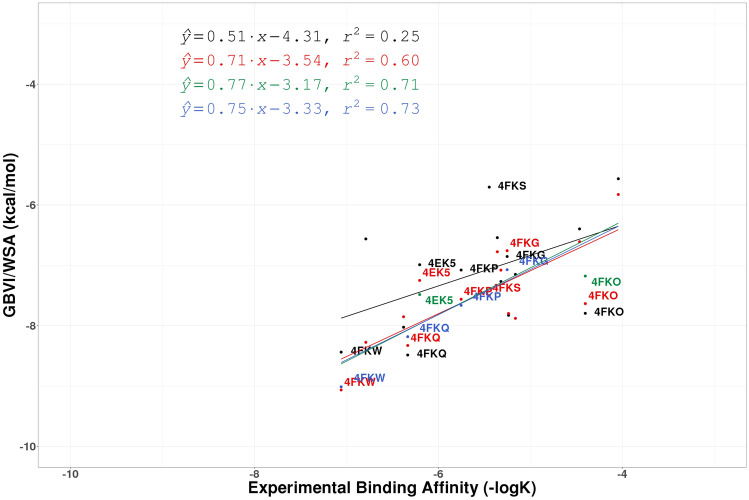
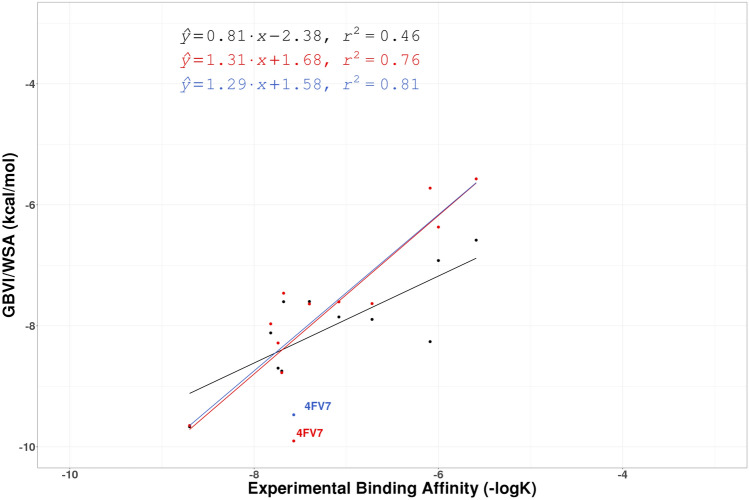
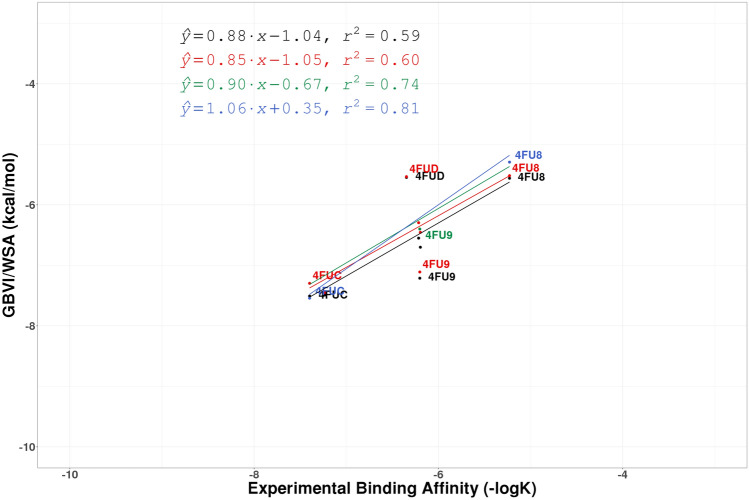
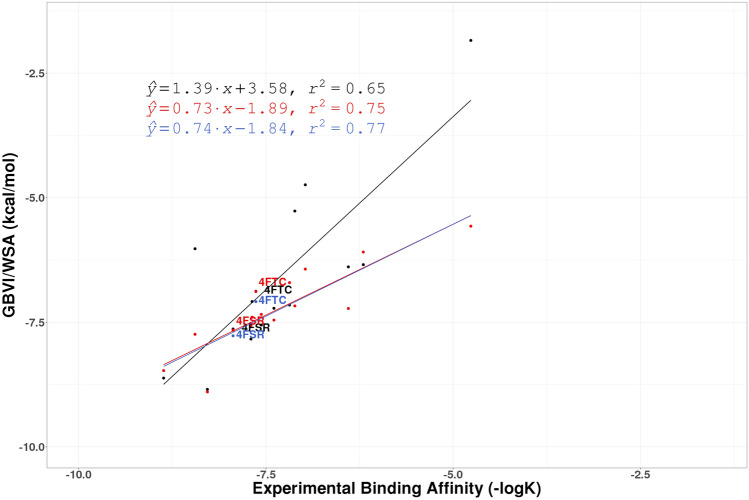
Similar articles
-
High-throughput quantum-mechanics/molecular-mechanics (ONIOM) macromolecular crystallographic refinement with PHENIX/DivCon: the impact of mixed Hamiltonian methods on ligand and protein structure.Acta Crystallogr D Struct Biol. 2018 Nov 1;74(Pt 11):1063-1077. doi: 10.1107/S2059798318012913. Epub 2018 Oct 29. Acta Crystallogr D Struct Biol. 2018. PMID: 30387765 Free PMC article.
-
Accurate macromolecular crystallographic refinement: incorporation of the linear scaling, semiempirical quantum-mechanics program DivCon into the PHENIX refinement package.Acta Crystallogr D Biol Crystallogr. 2014 May;70(Pt 5):1233-47. doi: 10.1107/S1399004714002260. Epub 2014 Apr 26. Acta Crystallogr D Biol Crystallogr. 2014. PMID: 24816093 Free PMC article.
-
XModeScore: a novel method for accurate protonation/tautomer-state determination using quantum-mechanically driven macromolecular X-ray crystallographic refinement.Acta Crystallogr D Struct Biol. 2016 Apr;72(Pt 4):586-98. doi: 10.1107/S2059798316002837. Epub 2016 Mar 30. Acta Crystallogr D Struct Biol. 2016. PMID: 27050137 Free PMC article.
-
Combining crystallography with quantum mechanics.Curr Opin Struct Biol. 2022 Feb;72:18-26. doi: 10.1016/j.sbi.2021.07.002. Epub 2021 Aug 12. Curr Opin Struct Biol. 2022. PMID: 34392061 Review.
-
QM Implementation in Drug Design: Does It Really Help?Methods Mol Biol. 2020;2114:19-35. doi: 10.1007/978-1-0716-0282-9_2. Methods Mol Biol. 2020. PMID: 32016884 Review.
Cited by
-
Favipiravir-Tautomeric and Complexation Properties in Solution.Pharmaceuticals (Basel). 2022 Dec 28;16(1):45. doi: 10.3390/ph16010045. Pharmaceuticals (Basel). 2022. PMID: 36678542 Free PMC article.
-
Refinement of RNA Structures Using Amber Force Fields.Crystals (Basel). 2021 Jul;11(7):771. doi: 10.3390/cryst11070771. Epub 2021 Jul 1. Crystals (Basel). 2021. PMID: 34745655 Free PMC article.
References
-
- Muller-Dethlefs K, Hobza P. Noncovalent interactions: a challenge for experiment and theory. Chem Rev. 2000;100:143–168. - PubMed
-
- Riley KE, Pitonak M, Cerny J, Hobza P. On the structure and geometry of biomolecular binding motifs (hydrogen-bonding, Stacking, X-H...pi): WFT and DFT calculations. J Chem Theory Comput. 2010;6:66–80. - PubMed
-
- Raha K, Peters MB, Wang B, Yu N, WollaCott AM, Westerhoff LM, Merz KM. The role of quantum mechanics in structure-based drug design. Drug Discov Today. 2007;12:725–731. - PubMed
-
- Kuntz ID. Structure-based strategies for drug design and discovery. Science. 1992;257:1078. - PubMed
-
- Jorgensen WL. The many roles of computation in drug discovery. Science. 2004;303:1813. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
