

Impaired capillary-to-arteriolar electrical signaling after traumatic brain injury
- PMID: 33050826
- PMCID: PMC8142130
- DOI: 10.1177/0271678X20962594
Impaired capillary-to-arteriolar electrical signaling after traumatic brain injury
Abstract
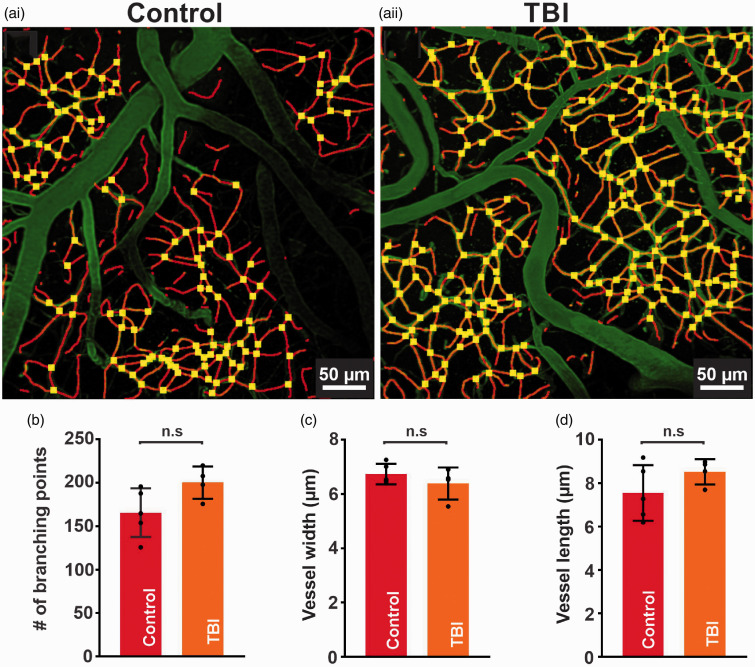
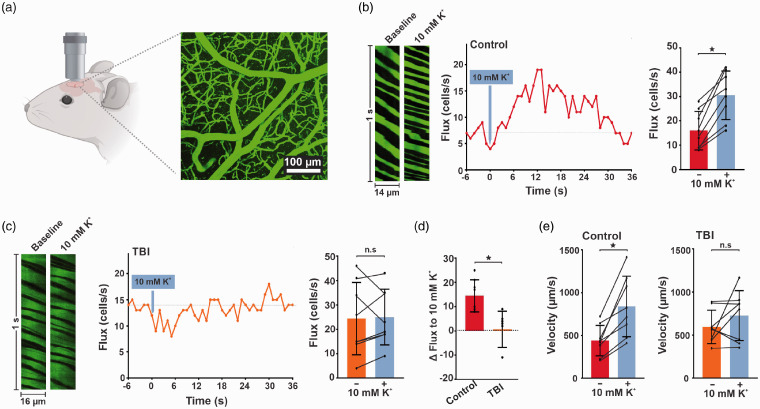
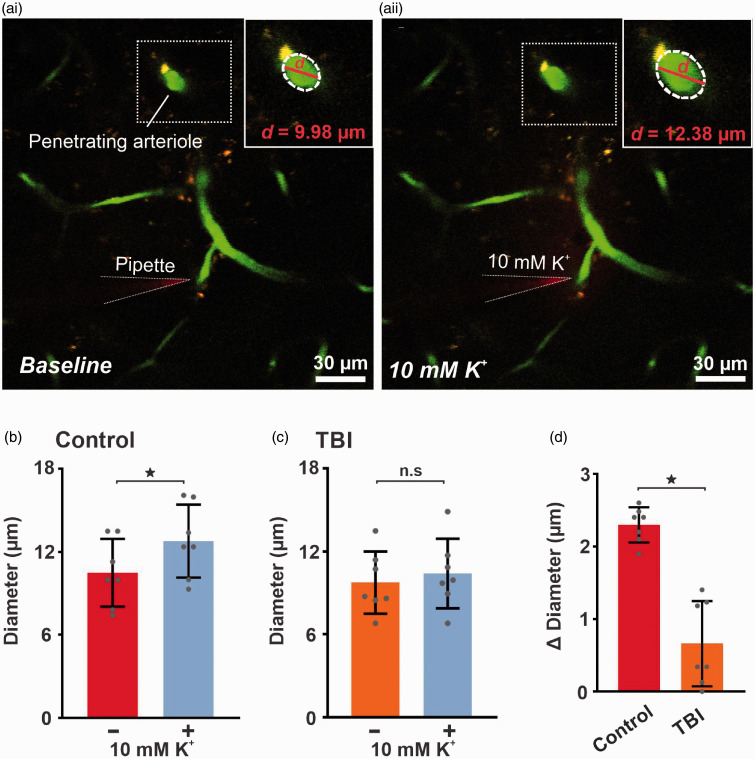
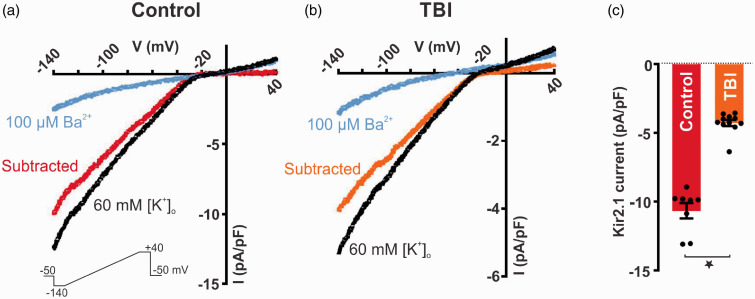
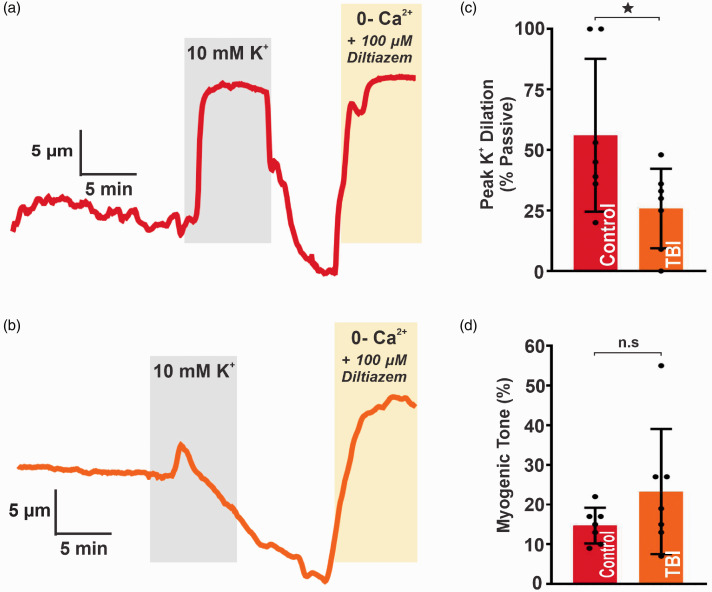
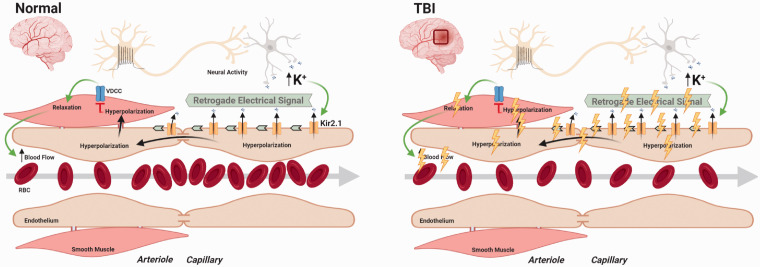
Traumatic brain injury (TBI) acutely impairs dynamic regulation of local cerebral blood flow, but long-term (>72 h) effects on functional hyperemia are unknown. Functional hyperemia depends on capillary endothelial cell inward rectifier potassium channels (Kir2.1) responding to potassium (K+) released during neuronal activity to produce a regenerative, hyperpolarizing electrical signal that propagates from capillaries to dilate upstream penetrating arterioles. We hypothesized that TBI causes widespread disruption of electrical signaling from capillaries-to-arterioles through impairment of Kir2.1 channel function. We randomized mice to TBI or control groups and allowed them to recover for 4 to 7 days post-injury. We measured in vivo cerebral hemodynamics and arteriolar responses to local stimulation of capillaries with 10 mM K+ using multiphoton laser scanning microscopy through a cranial window under urethane and α-chloralose anesthesia. Capillary angio-architecture was not significantly affected following injury. However, K+-induced hyperemia was significantly impaired. Electrophysiology recordings in freshly isolated capillary endothelial cells revealed diminished Ba2+-sensitive Kir2.1 currents, consistent with a reduction in channel function. In pressurized cerebral arteries isolated from TBI mice, K+ failed to elicit the vasodilation seen in controls. We conclude that disruption of endothelial Kir2.1 channel function impairs capillary-to-arteriole electrical signaling, contributing to altered cerebral hemodynamics after TBI.
Keywords: Cerebral blood flow; capillary endothelial cells; functional hyperemia; inward rectifier K+ channels (Kir2.1); traumatic brain injury.
Conflict of interest statement
Figures
Similar articles
-
Traumatic Brain Injury Impairs Systemic Vascular Function Through Disruption of Inward-Rectifier Potassium Channels.Function (Oxf). 2021;2(3):zqab018. doi: 10.1093/function/zqab018. Epub 2021 Apr 6. Function (Oxf). 2021. PMID: 34568829 Free PMC article.
-
Endothelial GqPCR activity controls capillary electrical signaling and brain blood flow through PIP2 depletion.Proc Natl Acad Sci U S A. 2018 Apr 10;115(15):E3569-E3577. doi: 10.1073/pnas.1800201115. Epub 2018 Mar 26. Proc Natl Acad Sci U S A. 2018. PMID: 29581272 Free PMC article.
-
PIP2 Improves Cerebral Blood Flow in a Mouse Model of Alzheimer's Disease.Function (Oxf). 2021 Feb 22;2(2):zqab010. doi: 10.1093/function/zqab010. eCollection 2021. Function (Oxf). 2021. PMID: 33763649 Free PMC article.
-
Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow.Microcirculation. 2015 Apr;22(3):183-96. doi: 10.1111/micc.12190. Microcirculation. 2015. PMID: 25641345 Free PMC article. Review.
-
Endothelial signaling at the core of neurovascular coupling: The emerging role of endothelial inward-rectifier K+ (Kir2.1) channels and N-methyl-d-aspartate receptors in the regulation of cerebral blood flow.Int J Biochem Cell Biol. 2021 Jun;135:105983. doi: 10.1016/j.biocel.2021.105983. Epub 2021 Apr 21. Int J Biochem Cell Biol. 2021. PMID: 33894355 Review.
Cited by
-
Cerebral arteriolar and neurovascular dysfunction after chemically induced menopause in mice.Am J Physiol Heart Circ Physiol. 2022 Nov 1;323(5):H845-H860. doi: 10.1152/ajpheart.00276.2022. Epub 2022 Sep 23. Am J Physiol Heart Circ Physiol. 2022. PMID: 36149767 Free PMC article.
-
Targeting hydrogen sulfide and nitric oxide to repair cardiovascular injury after trauma.Nitric Oxide. 2022 Dec 1;129:82-101. doi: 10.1016/j.niox.2022.10.003. Epub 2022 Oct 22. Nitric Oxide. 2022. PMID: 36280191 Free PMC article. Review.
-
Pathogenic soluble tau peptide disrupts endothelial calcium signaling and vasodilation in the brain microvasculature.J Cereb Blood Flow Metab. 2024 May;44(5):680-688. doi: 10.1177/0271678X241235790. Epub 2024 Feb 29. J Cereb Blood Flow Metab. 2024. PMID: 38420777
-
Endothelial KIR2 channel dysfunction in aged cerebral parenchymal arterioles.Am J Physiol Heart Circ Physiol. 2023 Oct 6;325(6):H1360-72. doi: 10.1152/ajpheart.00279.2023. Online ahead of print. Am J Physiol Heart Circ Physiol. 2023. PMID: 37801044 Free PMC article.
-
Traumatic Brain Injury Impairs Systemic Vascular Function Through Disruption of Inward-Rectifier Potassium Channels.Function (Oxf). 2021;2(3):zqab018. doi: 10.1093/function/zqab018. Epub 2021 Apr 6. Function (Oxf). 2021. PMID: 34568829 Free PMC article.
References
-
- Dewan MC, Rattani A, Gupta S, et al.. Estimating the global incidence of traumatic brain injury. J Neurosurg 2018; 1: 1–18. - PubMed
-
- Marin JR, Weaver MD, Yealy DM, et al.. Trends in visits for traumatic brain injury to emergency departments in the United States. JAMA 2014; 311: 1917–1919. - PubMed
-
- Abdul-Muneer PM, Conte AA, Haldar D, et al.. Traumatic brain injury induced matrix metalloproteinase2 cleaves CXCL12alpha (stromal cell derived factor 1alpha) and causes neurodegeneration. Brain Behav Immun 2017; 59: 190–199. - PubMed
-
- Roozenbeek B, Maas AI, Menon DK.Changing patterns in the epidemiology of traumatic brain injury. Nat Rev Neurol 2013; 9: 231–236. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
