

Predicting 3D genome folding from DNA sequence with Akita
- PMID: 33046897
- PMCID: PMC8211359
- DOI: 10.1038/s41592-020-0958-x
Predicting 3D genome folding from DNA sequence with Akita
Abstract
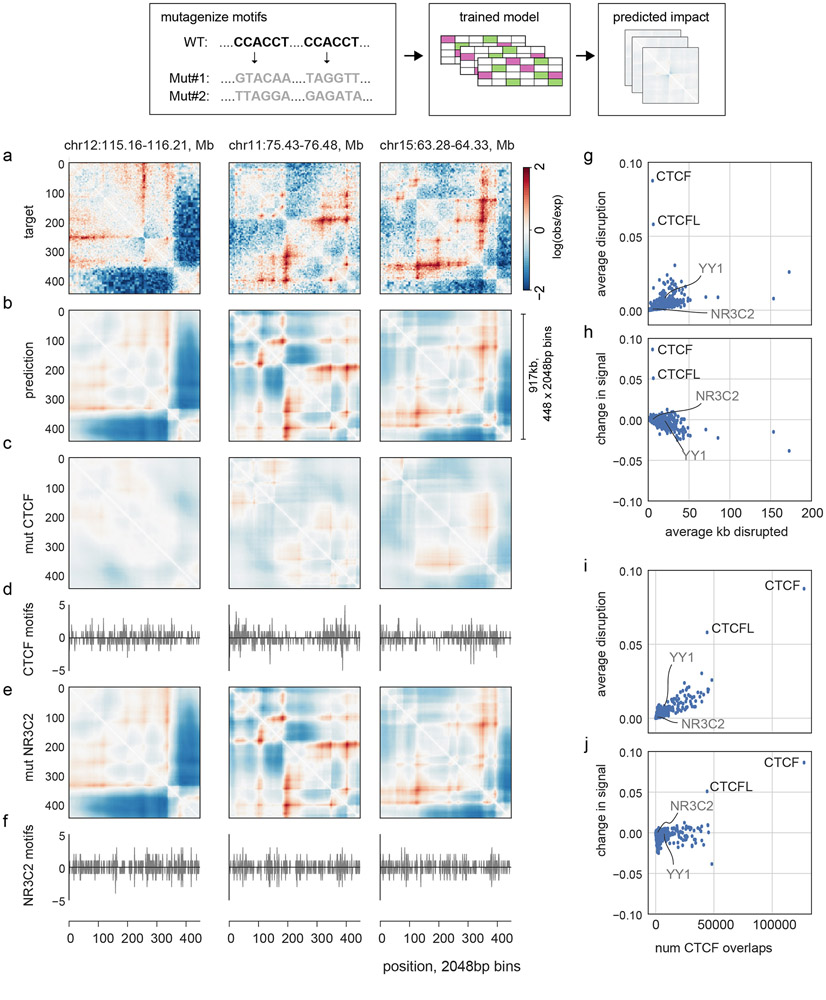
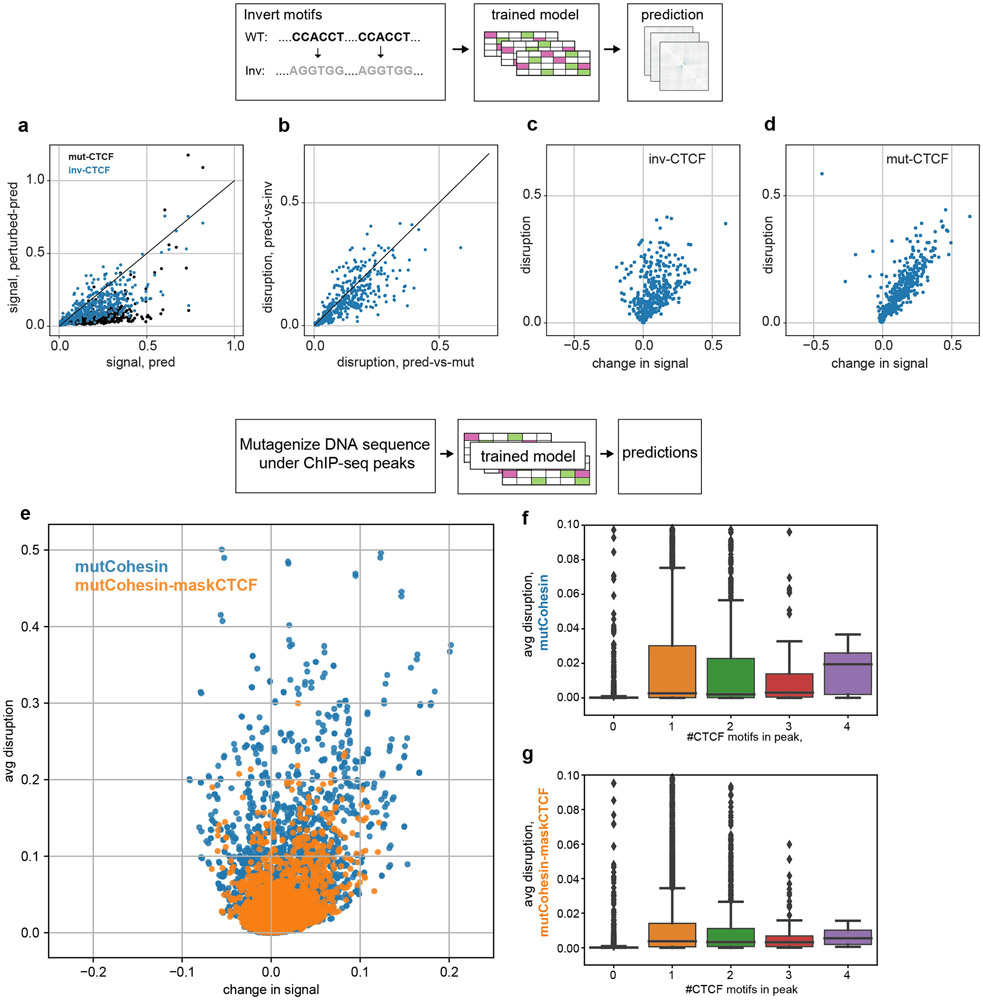
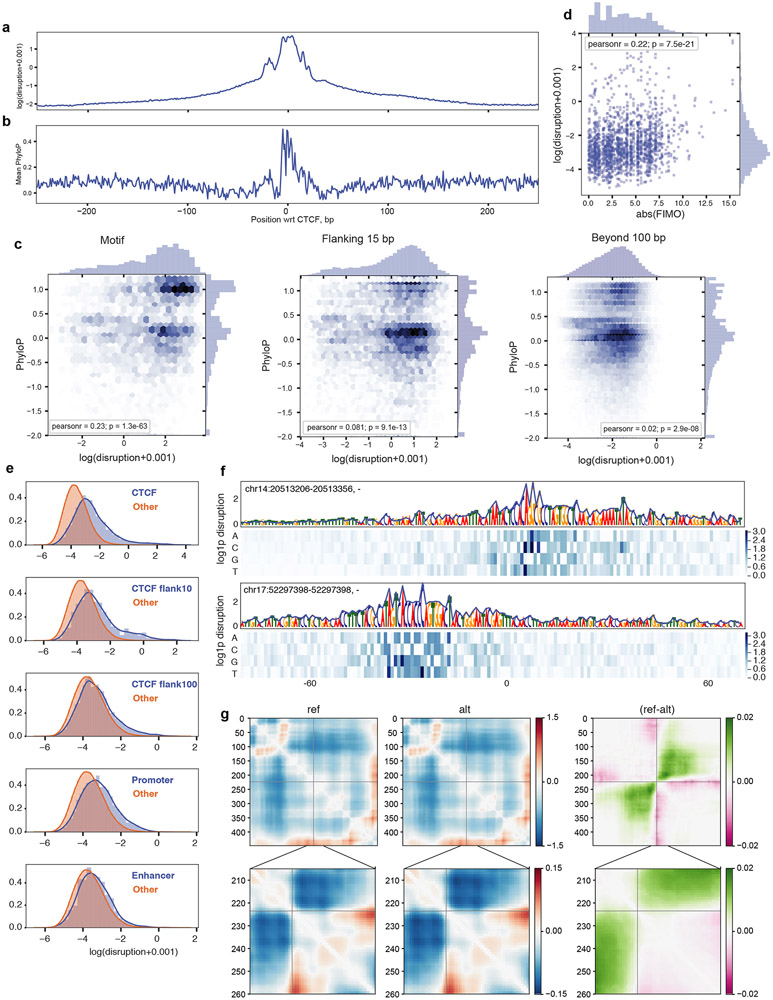
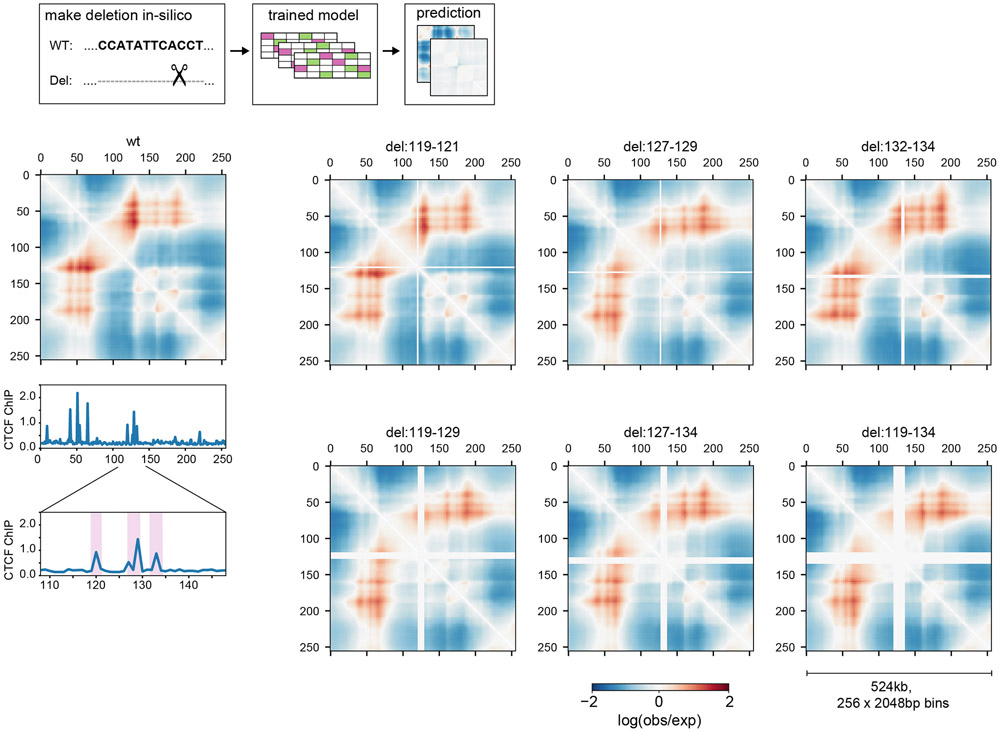
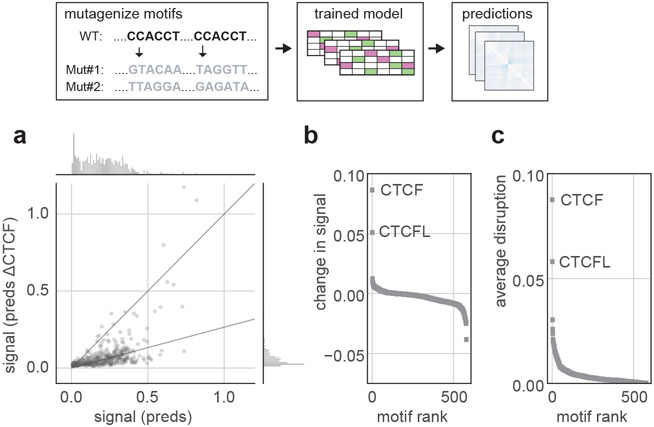
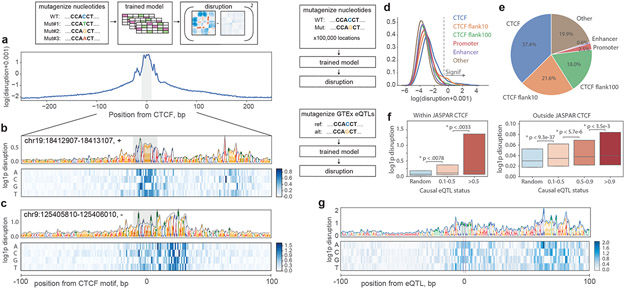
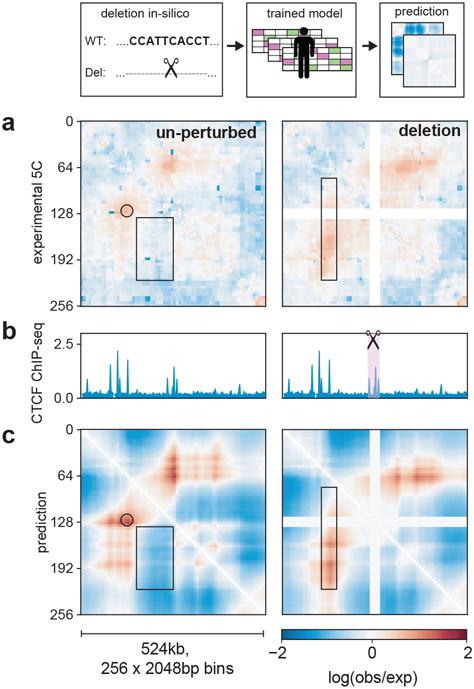
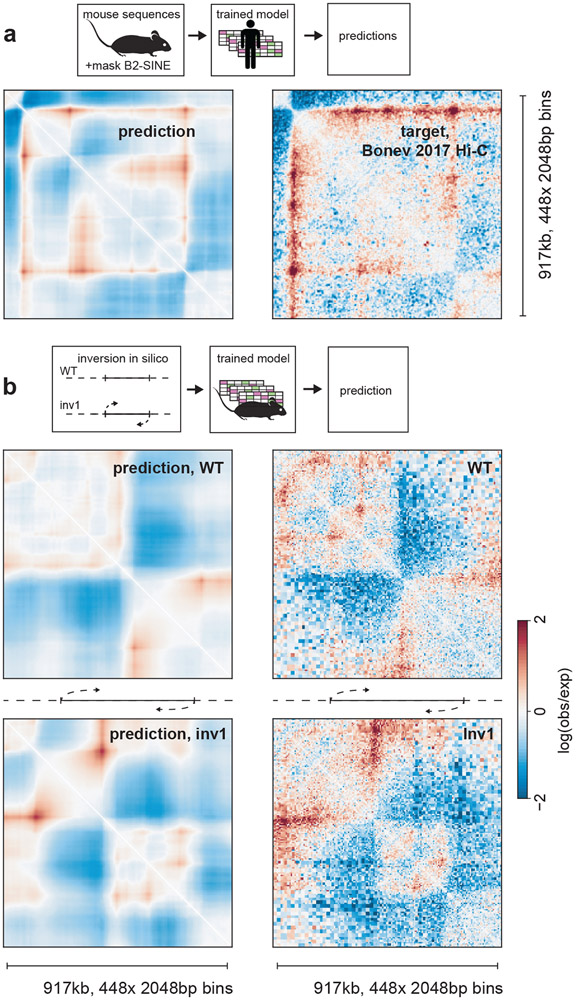
In interphase, the human genome sequence folds in three dimensions into a rich variety of locus-specific contact patterns. Cohesin and CTCF (CCCTC-binding factor) are key regulators; perturbing the levels of either greatly disrupts genome-wide folding as assayed by chromosome conformation capture methods. Still, how a given DNA sequence encodes a particular locus-specific folding pattern remains unknown. Here we present a convolutional neural network, Akita, that accurately predicts genome folding from DNA sequence alone. Representations learned by Akita underscore the importance of an orientation-specific grammar for CTCF binding sites. Akita learns predictive nucleotide-level features of genome folding, revealing effects of nucleotides beyond the core CTCF motif. Once trained, Akita enables rapid in silico predictions. Accounting for this, we demonstrate how Akita can be used to perform in silico saturation mutagenesis, interpret eQTLs, make predictions for structural variants and probe species-specific genome folding. Collectively, these results enable decoding genome function from sequence through structure.
Figures
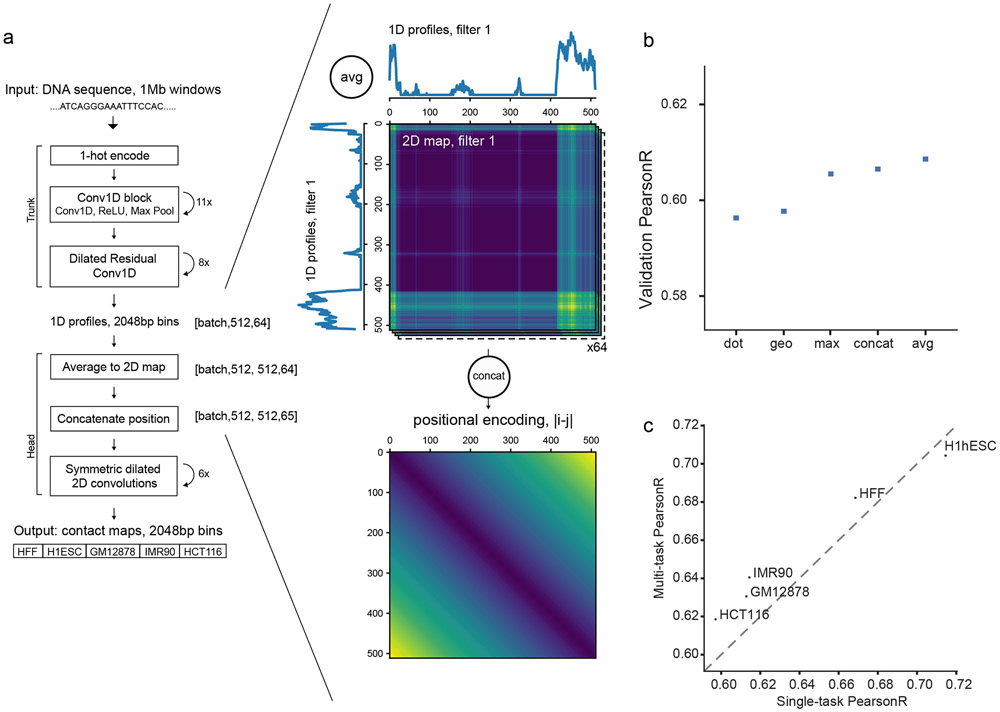
“dot”: Element-wise multiplication between each vector position, t(i,j,k) = oi(k)oj(k).
“geo”: Addition of one to all vector values, element-wise multiplication between each position, square root of each position, subtraction of one from all vector values,
“max”: Element-wise max between each vector position, t(i,j,k) = max(oi(k),oj(k)).
“concat”: Concatenate the two vectors, t(i,j) = [oi,oj].
“avg”: Element-wise mean between each vector position, t(i,j,k) = (oi(k)+oj(k))/2
Similar articles
-
CTCF and Cohesin in Genome Folding and Transcriptional Gene Regulation.Annu Rev Genomics Hum Genet. 2016 Aug 31;17:17-43. doi: 10.1146/annurev-genom-083115-022339. Epub 2016 Apr 18. Annu Rev Genomics Hum Genet. 2016. PMID: 27089971 Review.
-
Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders.Genome Biol. 2016 Aug 31;17(1):182. doi: 10.1186/s13059-016-1043-8. Genome Biol. 2016. PMID: 27582050 Free PMC article.
-
CTCF mediates chromatin looping via N-terminal domain-dependent cohesin retention.Proc Natl Acad Sci U S A. 2020 Jan 28;117(4):2020-2031. doi: 10.1073/pnas.1911708117. Epub 2020 Jan 14. Proc Natl Acad Sci U S A. 2020. PMID: 31937660 Free PMC article.
-
CTCF: a Swiss-army knife for genome organization and transcription regulation.Essays Biochem. 2019 Apr 23;63(1):157-165. doi: 10.1042/EBC20180069. Print 2019 Apr 23. Essays Biochem. 2019. PMID: 30940740 Review.
-
Genome-wide and parental allele-specific analysis of CTCF and cohesin DNA binding in mouse brain reveals a tissue-specific binding pattern and an association with imprinted differentially methylated regions.Genome Res. 2013 Oct;23(10):1624-35. doi: 10.1101/gr.150136.112. Epub 2013 Jun 26. Genome Res. 2013. PMID: 23804403 Free PMC article.
Cited by
-
Modeling transcriptional regulation of model species with deep learning.Genome Res. 2021 Jun;31(6):1097-1105. doi: 10.1101/gr.266171.120. Epub 2021 Apr 22. Genome Res. 2021. PMID: 33888512 Free PMC article.
-
The impact and future of artificial intelligence in medical genetics and molecular medicine: an ongoing revolution.Funct Integr Genomics. 2024 Aug 16;24(4):138. doi: 10.1007/s10142-024-01417-9. Funct Integr Genomics. 2024. PMID: 39147901 Review.
-
Machine learning reveals the diversity of human 3D chromatin contact patterns.bioRxiv [Preprint]. 2023 Dec 23:2023.12.22.573104. doi: 10.1101/2023.12.22.573104. bioRxiv. 2023. Update in: Mol Biol Evol. 2024 Oct 4;41(10):msae209. doi: 10.1093/molbev/msae209 PMID: 38187606 Free PMC article. Updated. Preprint.
-
Cross-species regulatory sequence activity prediction.PLoS Comput Biol. 2020 Jul 20;16(7):e1008050. doi: 10.1371/journal.pcbi.1008050. eCollection 2020 Jul. PLoS Comput Biol. 2020. PMID: 32687525 Free PMC article.
-
GenomicLinks: deep learning predictions of 3D chromatin interactions in the maize genome.NAR Genom Bioinform. 2024 Sep 24;6(3):lqae123. doi: 10.1093/nargab/lqae123. eCollection 2024 Sep. NAR Genom Bioinform. 2024. PMID: 39318505 Free PMC article.
References
-
- Merkenschlager M & Nora EP CTCF and Cohesin in Genome Folding and Transcriptional Gene Regulation. Annu. Rev. Genomics Hum. Genet 17, 17–43 (2016). - PubMed
-
- Krijger PHL & de Laat W Regulation of disease-associated gene expression in the 3D genome. Nat. Rev. Mol. Cell Biol 17, 771–782 (2016). - PubMed
-
- Despang A et al. Functional dissection of the Sox9-Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nat. Genet 51, 1263–1271 (2019). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
