

Innate Immunity Effector Cells as Inflammatory Drivers of Cardiac Fibrosis
- PMID: 32998408
- PMCID: PMC7583949
- DOI: 10.3390/ijms21197165
Innate Immunity Effector Cells as Inflammatory Drivers of Cardiac Fibrosis
Abstract
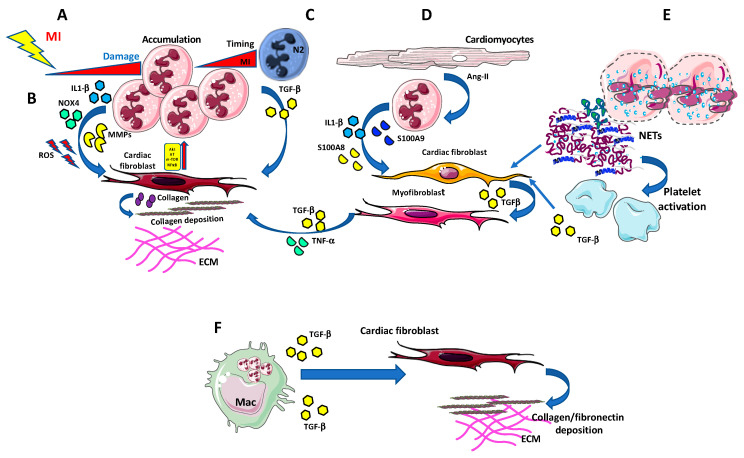
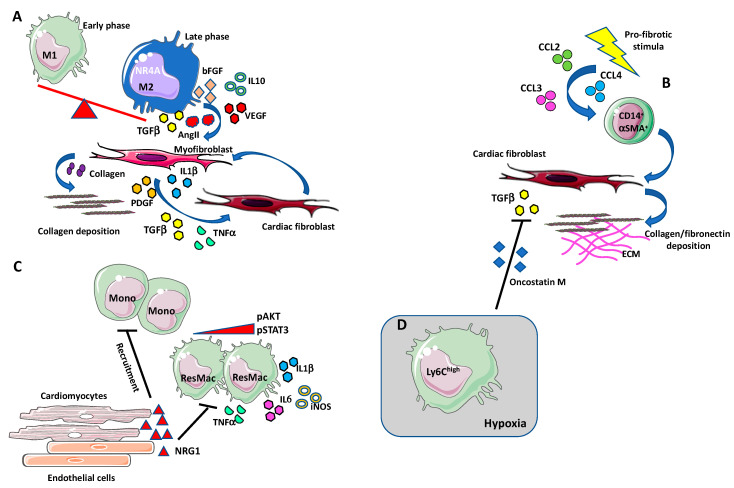
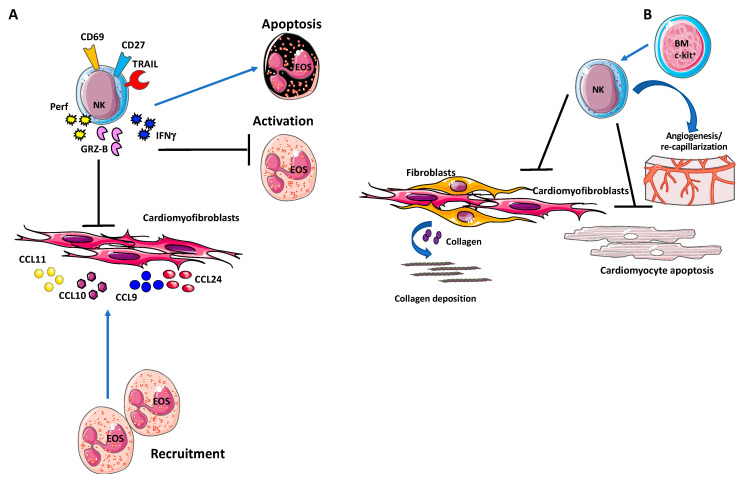
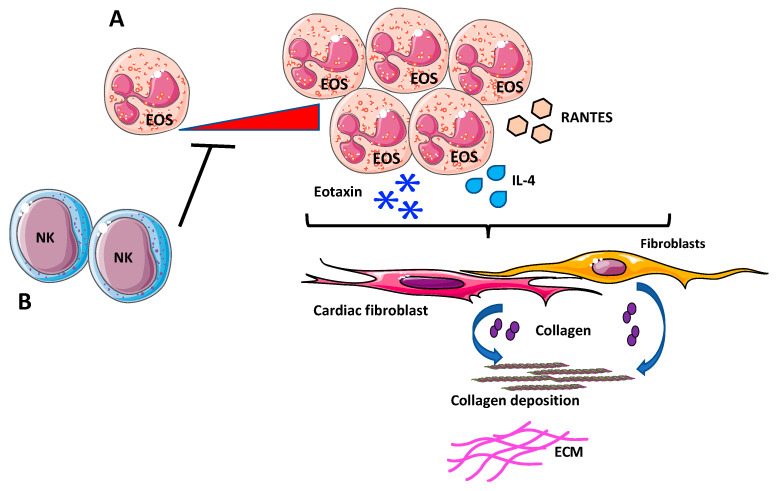
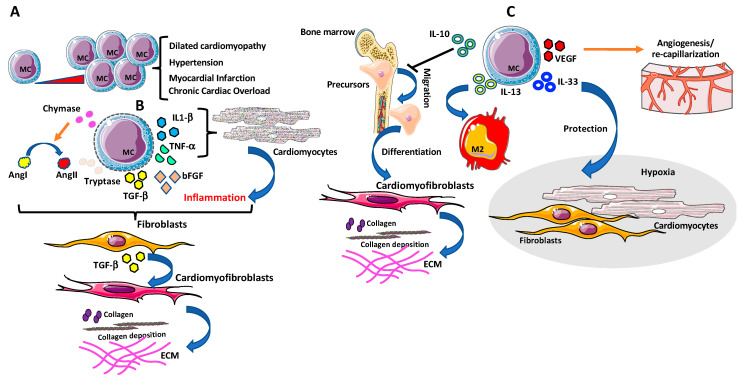
Despite relevant advances made in therapies for cardiovascular diseases (CVDs), they still represent the first cause of death worldwide. Cardiac fibrosis and excessive extracellular matrix (ECM) remodeling are common end-organ features in diseased hearts, leading to tissue stiffness, impaired myocardial functional, and progression to heart failure. Although fibrosis has been largely recognized to accompany and complicate various CVDs, events and mechanisms driving and governing fibrosis are still not entirely elucidated, and clinical interventions targeting cardiac fibrosis are not yet available. Immune cell types, both from innate and adaptive immunity, are involved not just in the classical response to pathogens, but they take an active part in "sterile" inflammation, in response to ischemia and other forms of injury. In this context, different cell types infiltrate the injured heart and release distinct pro-inflammatory cytokines that initiate the fibrotic response by triggering myofibroblast activation. The complex interplay between immune cells, fibroblasts, and other non-immune/host-derived cells is now considered as the major driving force of cardiac fibrosis. Here, we review and discuss the contribution of inflammatory cells of innate immunity, including neutrophils, macrophages, natural killer cells, eosinophils and mast cells, in modulating the myocardial microenvironment, by orchestrating the fibrogenic process in response to tissue injury. A better understanding of the time frame, sequences of events during immune cells infiltration, and their action in the injured inflammatory heart environment, may provide a rationale to design new and more efficacious therapeutic interventions to reduce cardiac fibrosis.
Keywords: cardiac fibrosis; eosinophils; inflammation; macrophages; mast cells; natural killer cells; neutrophils.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
T-cell regulation of fibroblasts and cardiac fibrosis.Matrix Biol. 2020 Sep;91-92:167-175. doi: 10.1016/j.matbio.2020.04.001. Epub 2020 May 11. Matrix Biol. 2020. PMID: 32438054 Free PMC article. Review.
-
Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities.Mol Aspects Med. 2019 Feb;65:70-99. doi: 10.1016/j.mam.2018.07.001. Epub 2018 Aug 2. Mol Aspects Med. 2019. PMID: 30056242 Review.
-
The pathogenesis of cardiac fibrosis.Cell Mol Life Sci. 2014 Feb;71(4):549-74. doi: 10.1007/s00018-013-1349-6. Epub 2013 May 7. Cell Mol Life Sci. 2014. PMID: 23649149 Free PMC article. Review.
-
A double-edged sword of immuno-microenvironment in cardiac homeostasis and injury repair.Signal Transduct Target Ther. 2021 Feb 22;6(1):79. doi: 10.1038/s41392-020-00455-6. Signal Transduct Target Ther. 2021. PMID: 33612829 Free PMC article. Review.
-
Immune cells as targets for cardioprotection: new players and novel therapeutic opportunities.Cardiovasc Res. 2019 Jun 1;115(7):1117-1130. doi: 10.1093/cvr/cvz050. Cardiovasc Res. 2019. PMID: 30825305 Free PMC article. Review.
Cited by
-
When a Friend Becomes Your Enemy: Natural Killer Cells in Atherosclerosis and Atherosclerosis-Associated Risk Factors.Front Immunol. 2022 Jan 13;12:798155. doi: 10.3389/fimmu.2021.798155. eCollection 2021. Front Immunol. 2022. PMID: 35095876 Free PMC article. Review.
-
High frequencies of adaptive NK cells are associated with absence of coronary plaque in cytomegalovirus infected people living with HIV.Medicine (Baltimore). 2022 Sep 23;101(38):e30794. doi: 10.1097/MD.0000000000030794. Medicine (Baltimore). 2022. PMID: 36197157 Free PMC article.
-
Exploring the causal relationship between immune cell and all-cause heart failure: a Mendelian randomization study.Front Cardiovasc Med. 2024 Jun 13;11:1363200. doi: 10.3389/fcvm.2024.1363200. eCollection 2024. Front Cardiovasc Med. 2024. PMID: 38938655 Free PMC article.
-
Cardiac Mast Cells: A Two-Head Regulator in Cardiac Homeostasis and Pathogenesis Following Injury.Front Immunol. 2022 Jul 15;13:963444. doi: 10.3389/fimmu.2022.963444. eCollection 2022. Front Immunol. 2022. PMID: 35911776 Free PMC article. Review.
-
The NLRP3 Inflammasome as a Novel Therapeutic Target for Cardiac Fibrosis.J Inflamm Res. 2022 Jul 7;15:3847-3858. doi: 10.2147/JIR.S370483. eCollection 2022. J Inflamm Res. 2022. PMID: 35836721 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
