

Mitochondrial dysfunction in fibrotic diseases
- PMID: 32963808
- PMCID: PMC7474731
- DOI: 10.1038/s41420-020-00316-9
Mitochondrial dysfunction in fibrotic diseases
Abstract
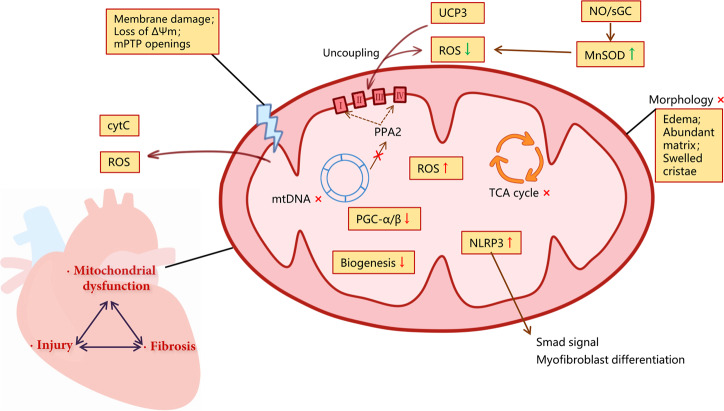
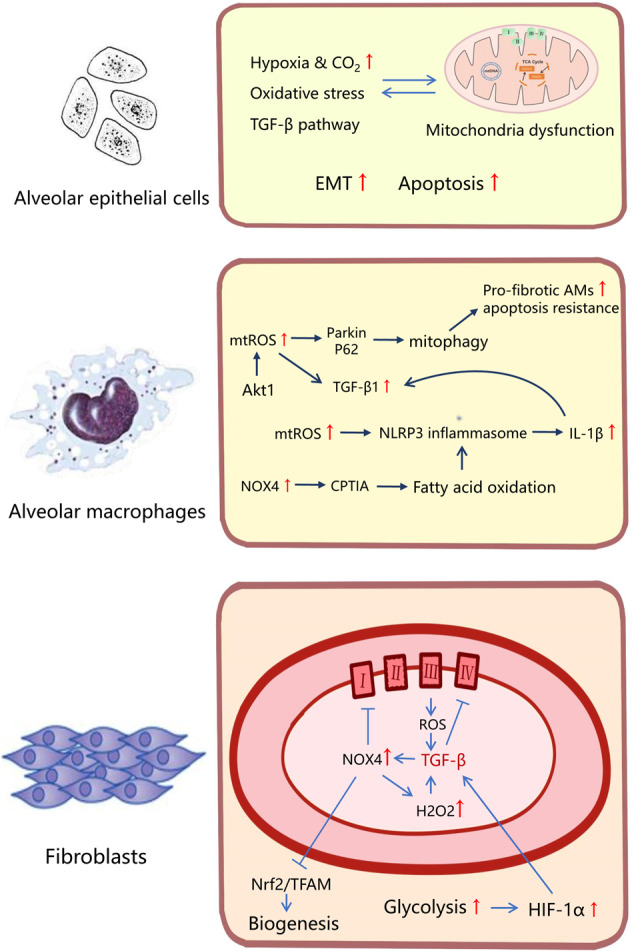
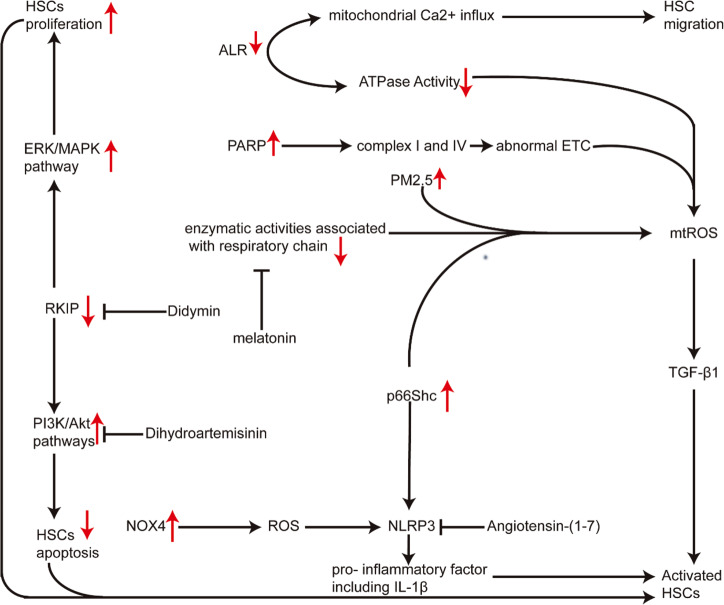
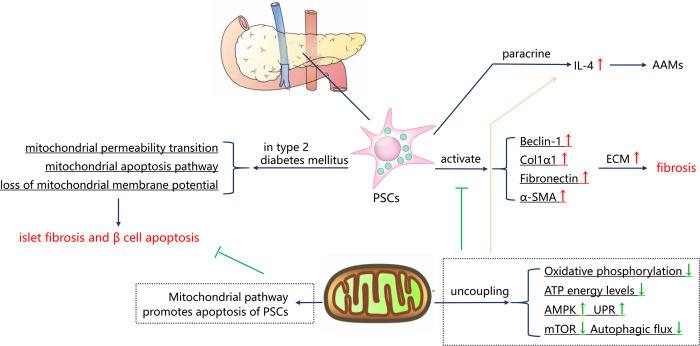
Although fibrosis is a common pathological feature of most end-stage organ diseases, its pathogenesis remains unclear. There is growing evidence that mitochondrial dysfunction contributes to the development and progression of fibrosis. The heart, liver, kidney and lung are highly oxygen-consuming organs that are sensitive to mitochondrial dysfunction. Moreover, the fibrotic process of skin and islet is closely related to mitochondrial dysfunction as well. This review summarized emerging mechanisms related to mitochondrial dysfunction in different fibrotic organs and tissues above. First, it highlighted the important elucidation of mitochondria morphological changes, mitochondrial membrane potential and structural damage, mitochondrial DNA (mtDNA) damage and reactive oxidative species (ROS) production, etc. Second, it introduced the abnormality of mitophagy and mitochondrial transfer also contributed to the fibrotic process. Therefore, with gaining the increasing knowledge of mitochondrial structure, function, and origin, we could kindle a new era for the diagnostic and therapeutic strategies of many fibrotic diseases based on mitochondrial dysfunction.
Keywords: Metabolic disorders; Mitochondria.
© The Author(s) 2020.
Conflict of interest statement
Conflict of interestThe authors declare that they have no conflict of interest.
Figures
Similar articles
-
Mitochondrial dysfunction in diabetic kidney disease.Clin Chim Acta. 2019 Sep;496:108-116. doi: 10.1016/j.cca.2019.07.005. Epub 2019 Jul 2. Clin Chim Acta. 2019. PMID: 31276635 Review.
-
Mitochondrial biogenesis: pharmacological approaches.Curr Pharm Des. 2014;20(35):5507-9. doi: 10.2174/138161282035140911142118. Curr Pharm Des. 2014. PMID: 24606795
-
Mitochondrial Quality Control in Age-Related Pulmonary Fibrosis.Int J Mol Sci. 2020 Jan 18;21(2):643. doi: 10.3390/ijms21020643. Int J Mol Sci. 2020. PMID: 31963720 Free PMC article. Review.
-
Mitochondrial destiny in type 2 diabetes: the effects of oxidative stress on the dynamics and biogenesis of mitochondria.PeerJ. 2020 Aug 25;8:e9741. doi: 10.7717/peerj.9741. eCollection 2020. PeerJ. 2020. PMID: 32904391 Free PMC article.
-
[Pathways for maintenance of mitochondrial DNA integrity and mitochondrial functions in cells exposed to ionizing radiation].Radiats Biol Radioecol. 2013 Mar-Apr;53(2):117-36. doi: 10.7868/s0869803113020045. Radiats Biol Radioecol. 2013. PMID: 23786028 Review. Russian.
Cited by
-
Microvesicles released from pneumolysin-stimulated lung epithelial cells carry mitochondrial cargo and suppress neutrophil oxidative burst.Sci Rep. 2021 May 5;11(1):9529. doi: 10.1038/s41598-021-88897-y. Sci Rep. 2021. PMID: 33953279 Free PMC article.
-
The Role and Mechanism of Oxidative Stress and Nuclear Receptors in the Development of NAFLD.Oxid Med Cell Longev. 2021 Oct 27;2021:6889533. doi: 10.1155/2021/6889533. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 34745420 Free PMC article. Review.
-
Targeting Pulmonary Fibrosis by SLC1A5-Dependent Glutamine Transport Blockade.Am J Respir Cell Mol Biol. 2023 Oct;69(4):441-455. doi: 10.1165/rcmb.2022-0339OC. Am J Respir Cell Mol Biol. 2023. PMID: 37459644 Free PMC article.
-
Aging-Related Mitochondrial Dysfunction Is Associated With Fibrosis in Benign Prostatic Hyperplasia.J Gerontol A Biol Sci Med Sci. 2024 Jun 1;79(6):glad222. doi: 10.1093/gerona/glad222. J Gerontol A Biol Sci Med Sci. 2024. PMID: 37738211 Free PMC article.
-
Mitochondrial Dysfunction in Heart Failure: From Pathophysiological Mechanisms to Therapeutic Opportunities.Int J Mol Sci. 2024 Feb 25;25(5):2667. doi: 10.3390/ijms25052667. Int J Mol Sci. 2024. PMID: 38473911 Free PMC article. Review.
References
-
- Humphreys BD. Mechanisms of renal fibrosis. Annu. Rev. Physiol. 2018;80:309–326. - PubMed
-
- Ge PS, Runyon BA. Treatment of patients with cirrhosis. N. Engl. J. Med. 2016;375:767–777. - PubMed
-
- Topf U, Wrobel L, Chacinska A. Chatty mitochondria: keeping balance in cellular protein homeostasis. Trends Cell Biol. 2016;26:577–586. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
