

Fanconi-Bickel Syndrome: A Review of the Mechanisms That Lead to Dysglycaemia
- PMID: 32877990
- PMCID: PMC7504390
- DOI: 10.3390/ijms21176286
Fanconi-Bickel Syndrome: A Review of the Mechanisms That Lead to Dysglycaemia
Abstract
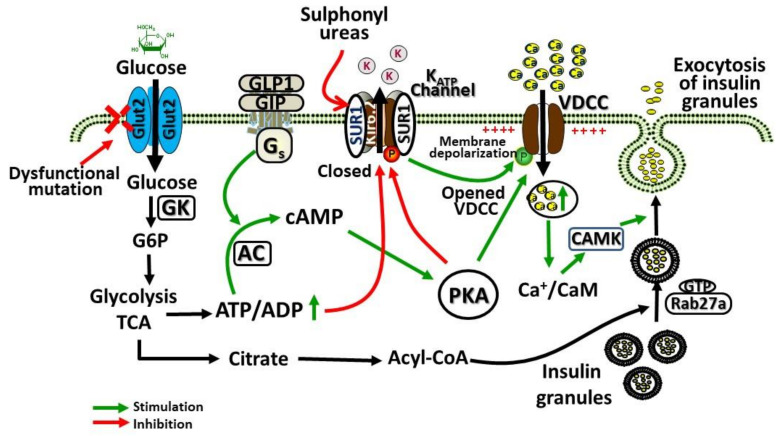
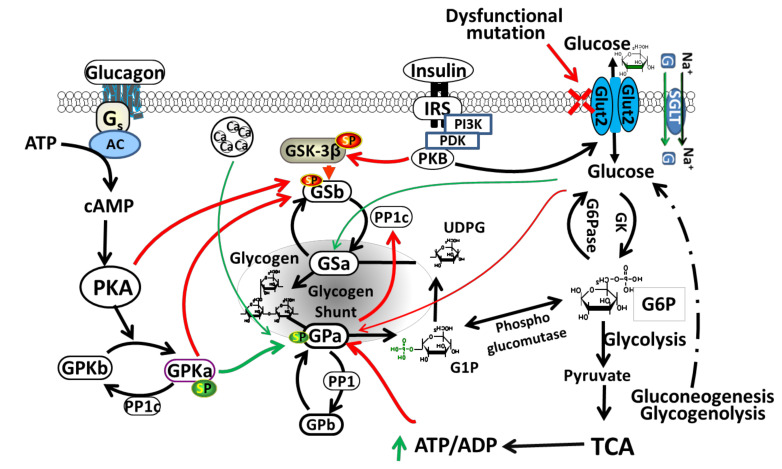
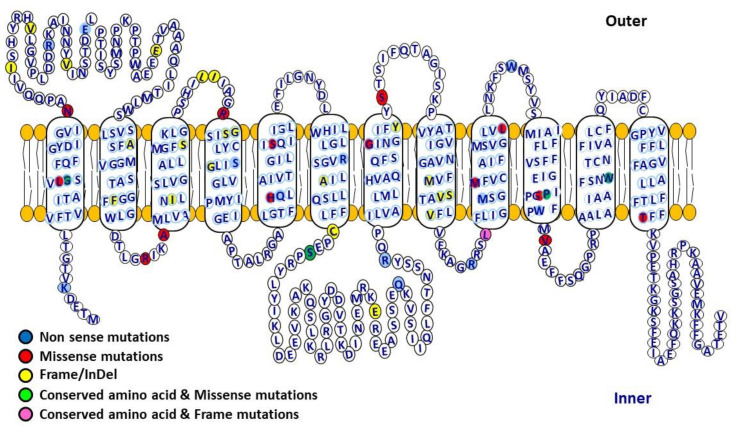
Accumulation of glycogen in the kidney and liver is the main feature of Fanconi-Bickel Syndrome (FBS), a rare disorder of carbohydrate metabolism inherited in an autosomal recessive manner due to SLC2A2 gene mutations. Missense, nonsense, frame-shift (fs), in-frame indels, splice site, and compound heterozygous variants have all been identified in SLC2A2 gene of FBS cases. Approximately 144 FBS cases with 70 different SLC2A2 gene variants have been reported so far. SLC2A2 encodes for glucose transporter 2 (GLUT2) a low affinity facilitative transporter of glucose mainly expressed in tissues playing important roles in glucose homeostasis, such as renal tubular cells, enterocytes, pancreatic β-cells, hepatocytes and discrete regions of the brain. Dysfunctional mutations and decreased GLUT2 expression leads to dysglycaemia (fasting hypoglycemia, postprandial hyperglycemia, glucose intolerance, and rarely diabetes mellitus), hepatomegaly, galactose intolerance, rickets, and poor growth. The molecular mechanisms of dysglycaemia in FBS are still not clearly understood. In this review, we discuss the physiological roles of GLUT2 and the pathophysiology of mutants, highlight all of the previously reported SLC2A2 mutations associated with dysglycaemia, and review the potential molecular mechanisms leading to dysglycaemia and diabetes mellitus in FBS patients.
Keywords: Fanconi–Bickel Syndrome (FBS); GLUT2 dysfunction; SLC2A2 mutation; birth weight; cAMP; dysglycaemia; hepatomegaly; insulin secretion; liver; pancreatic β cell.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Impaired glucose tolerance in Fanconi-Bickel syndrome: Eight patients with two novel mutations.Turk J Pediatr. 2017;59(4):434-441. doi: 10.24953/turkjped.2017.04.010. Turk J Pediatr. 2017. PMID: 29624224
-
Genetic testing of two Pakistani patients affected with rare autosomal recessive Fanconi-Bickel syndrome and identification of a novel SLC2A2 splice site variant.J Pediatr Endocrinol Metab. 2019 Nov 26;32(11):1229-1233. doi: 10.1515/jpem-2019-0235. J Pediatr Endocrinol Metab. 2019. PMID: 31473689
-
Mutation analysis of the GLUT2 gene in three unrelated Egyptian families with Fanconi-Bickel syndrome: revisited gene atlas for renumbering.Clin Exp Nephrol. 2012 Aug;16(4):604-10. doi: 10.1007/s10157-012-0603-9. Epub 2012 Feb 18. Clin Exp Nephrol. 2012. PMID: 22350464
-
Fanconi-Bickel syndrome--a congenital defect of facilitative glucose transport.Curr Mol Med. 2002 Mar;2(2):213-27. doi: 10.2174/1566524024605743. Curr Mol Med. 2002. PMID: 11949937 Review.
-
Fanconi-Bickel syndrome--the original patient and his natural history, historical steps leading to the primary defect, and a review of the literature.Eur J Pediatr. 1998 Oct;157(10):783-97. doi: 10.1007/s004310050937. Eur J Pediatr. 1998. PMID: 9809815 Review.
Cited by
-
Understanding the Mechanism of Dysglycemia in a Fanconi-Bickel Syndrome Patient.Front Endocrinol (Lausanne). 2022 May 18;13:841788. doi: 10.3389/fendo.2022.841788. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 35663312 Free PMC article.
-
Whole-Exome Sequencing Uncovers Novel Causative Variants and Additional Findings in Three Patients Affected by Glycogen Storage Disease Type VI and Fanconi-Bickel Syndrome.Front Genet. 2021 Jan 11;11:601566. doi: 10.3389/fgene.2020.601566. eCollection 2020. Front Genet. 2021. PMID: 33505429 Free PMC article.
-
Hypoglycaemia Metabolic Gene Panel Testing.Front Endocrinol (Lausanne). 2022 Mar 29;13:826167. doi: 10.3389/fendo.2022.826167. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 35422763 Free PMC article. Review.
-
Glycogen storage diseases: An update.World J Gastroenterol. 2023 Jul 7;29(25):3932-3963. doi: 10.3748/wjg.v29.i25.3932. World J Gastroenterol. 2023. PMID: 37476587 Free PMC article. Review.
-
Attenuated glucose uptake promotes catabolic metabolism through activated AMPK signaling and impaired insulin signaling in zebrafish.Front Nutr. 2023 May 26;10:1187283. doi: 10.3389/fnut.2023.1187283. eCollection 2023. Front Nutr. 2023. PMID: 37305084 Free PMC article.
References
-
- Fanconi G., Bickel H. [Chronic aminoaciduria (amino acid diabetes or nephrotic-glucosuric dwarfism) in glycogen storage and cystine disease] Helv. Paediatr. Acta. 1949;4:359–396. - PubMed
-
- Mueckler M., Kruse M., Strube M., Riggs A.C., Chiu K.C., Permutt M.A. A mutation in the Glut2 glucose transporter gene of a diabetic patient abolishes transport activity. J. Biol. Chem. 1994;269:17765–17767. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
