

G551D mutation impairs PKA-dependent activation of CFTR channel that can be restored by novel GOF mutations
- PMID: 32877225
- PMCID: PMC7701354
- DOI: 10.1152/ajplung.00262.2019
G551D mutation impairs PKA-dependent activation of CFTR channel that can be restored by novel GOF mutations
Abstract
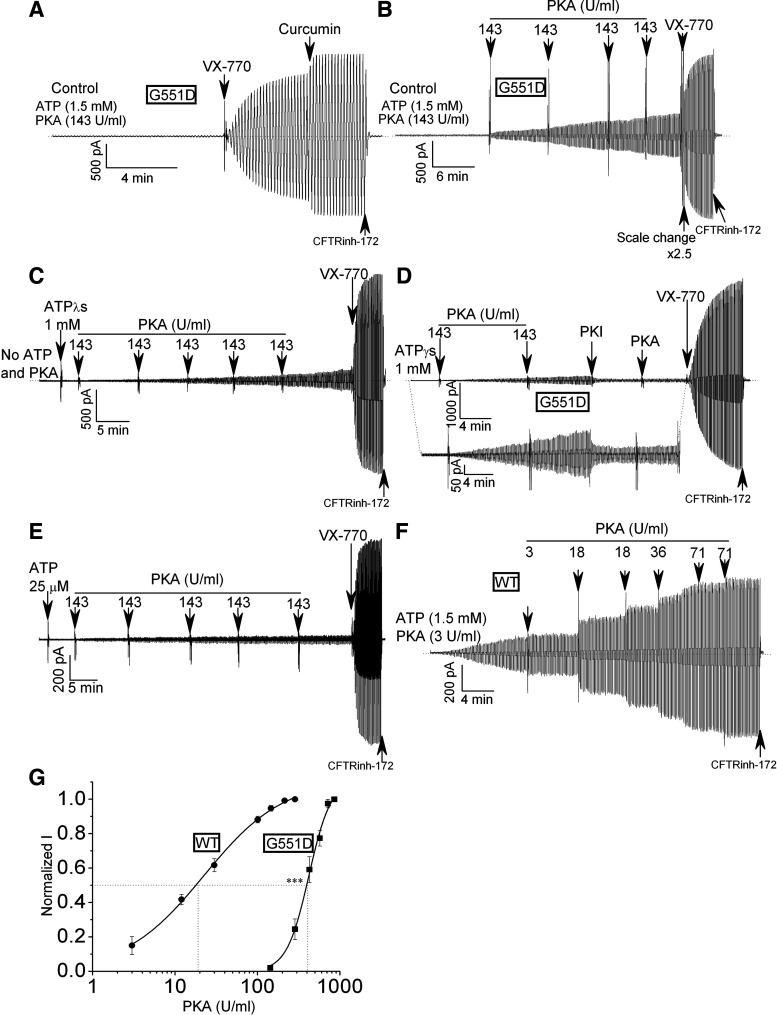
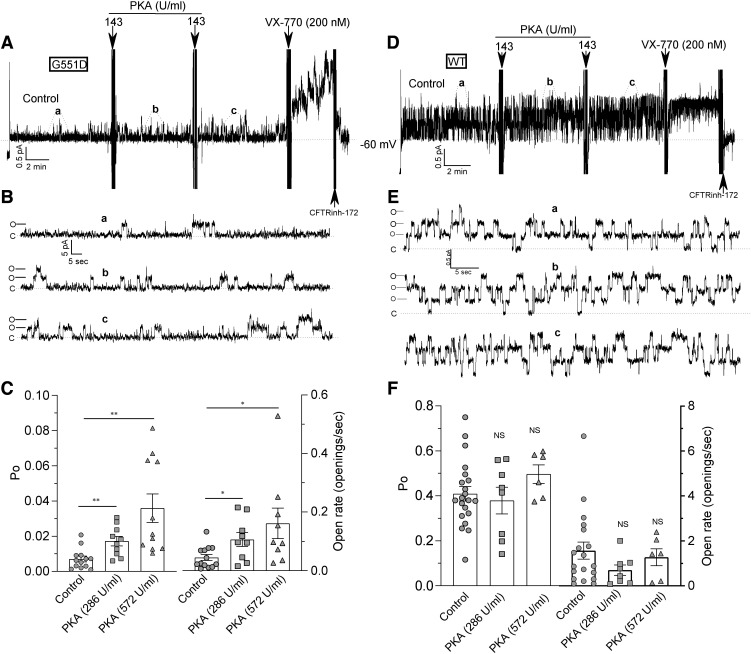
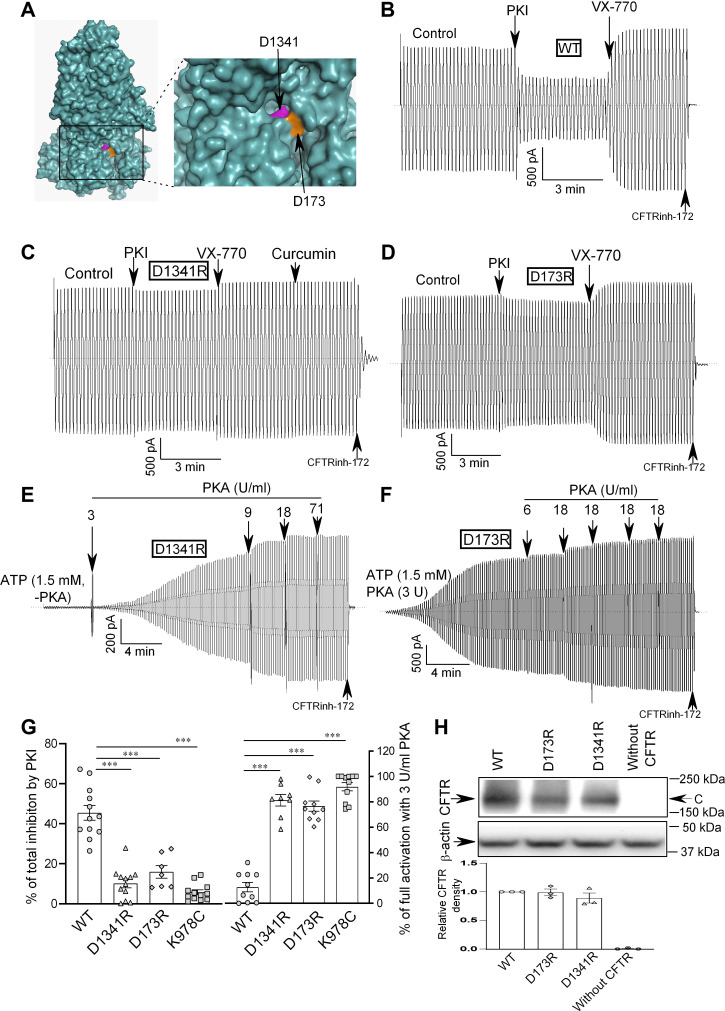
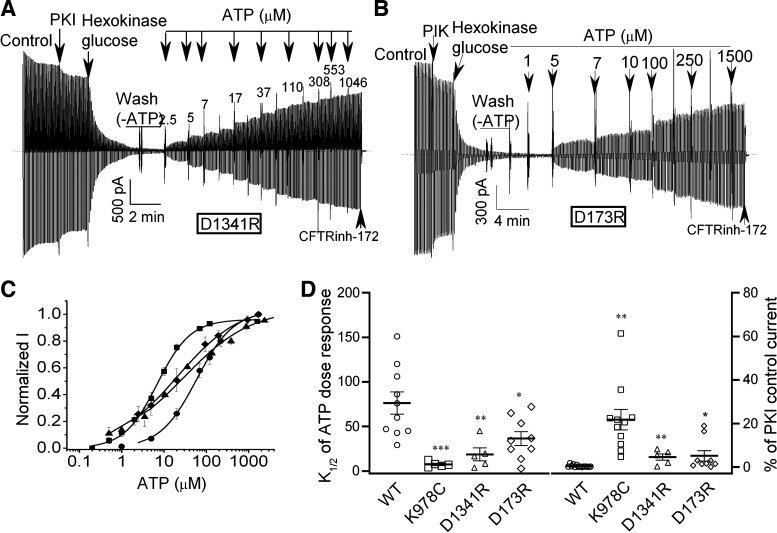
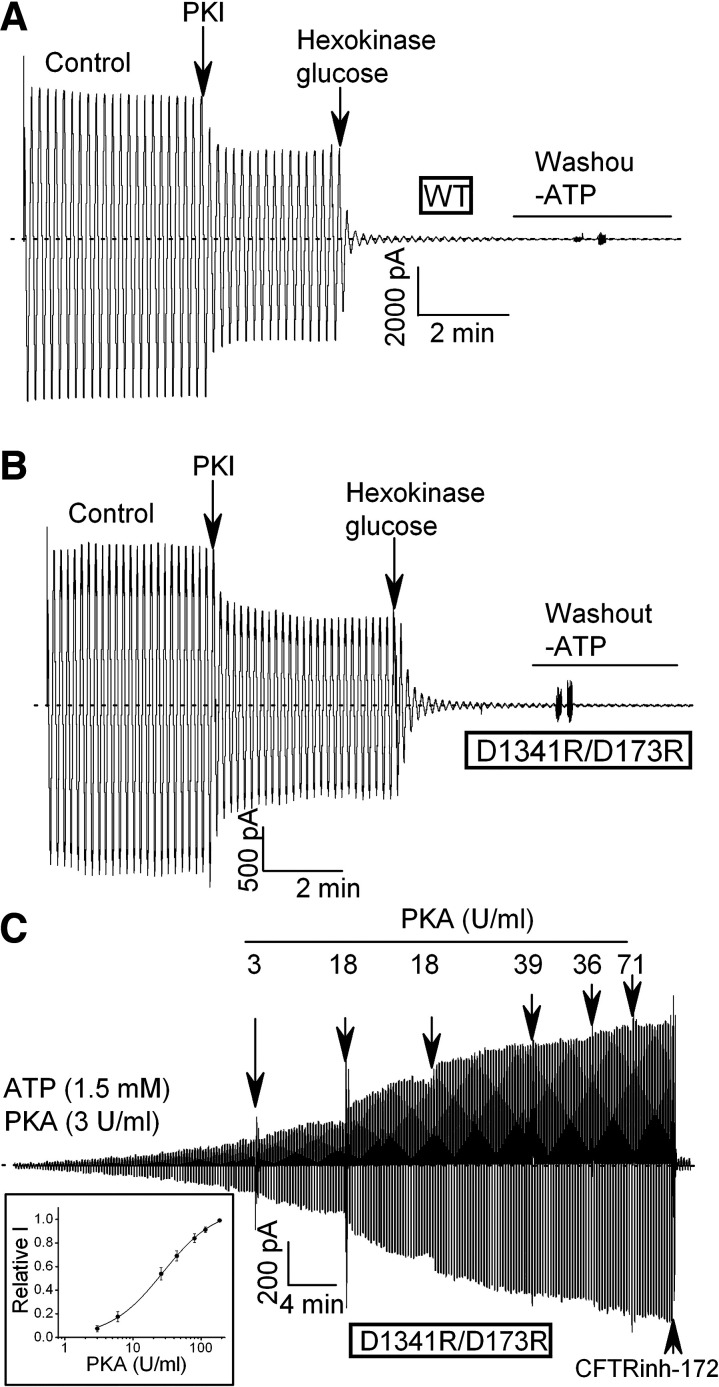
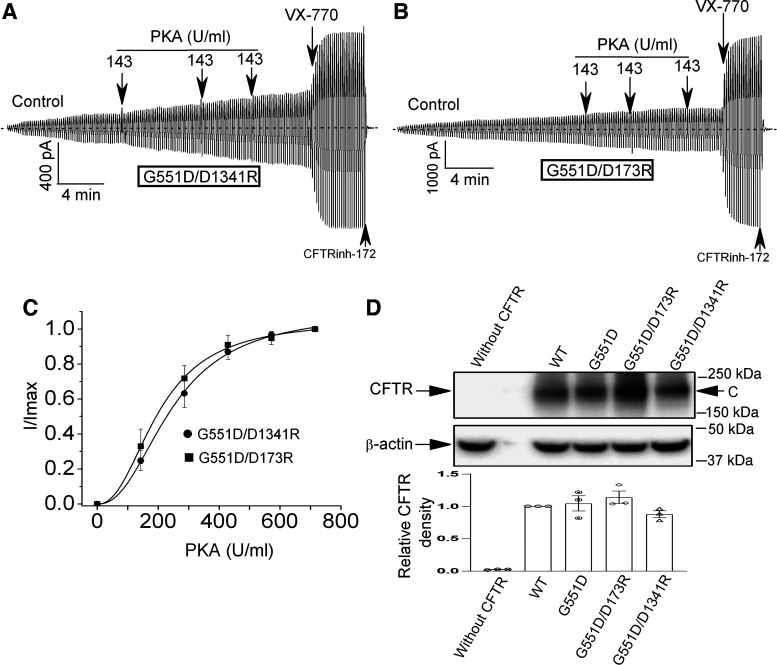
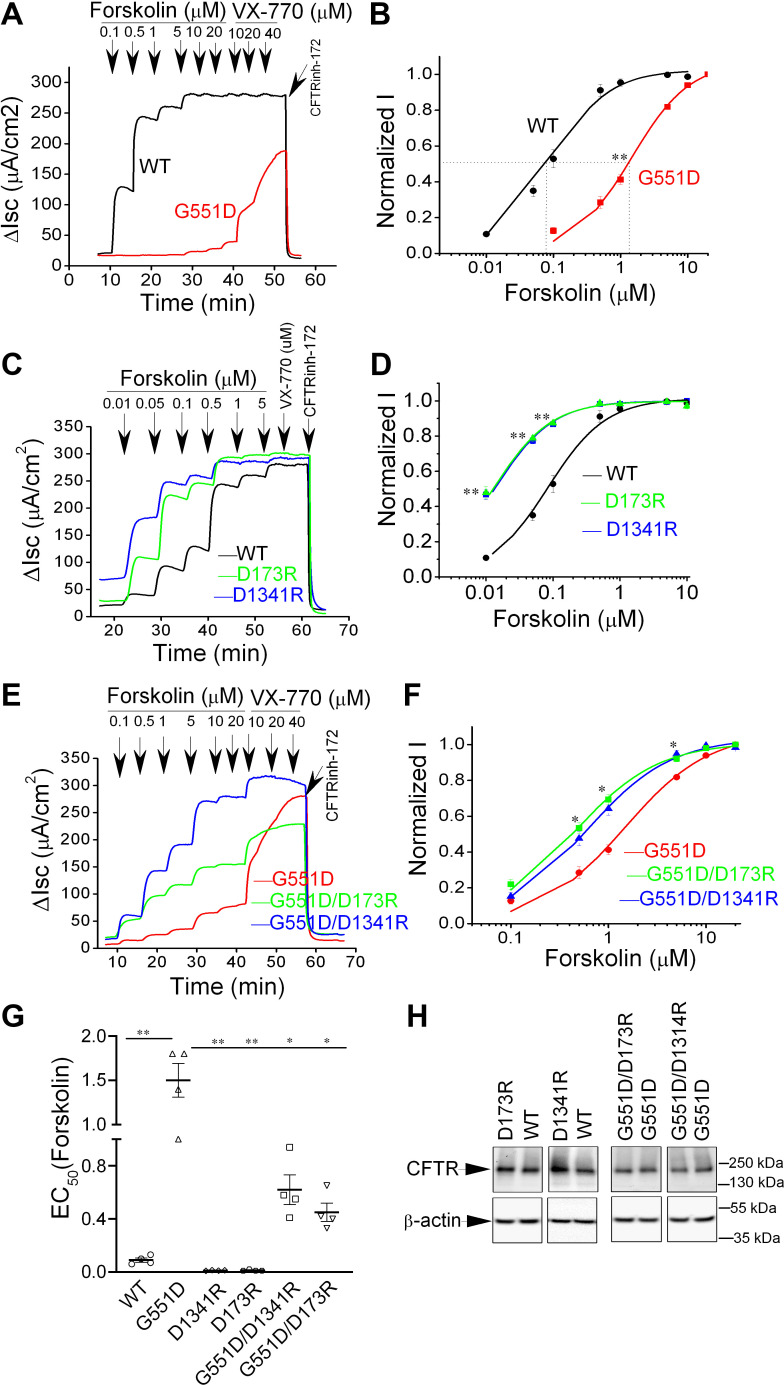
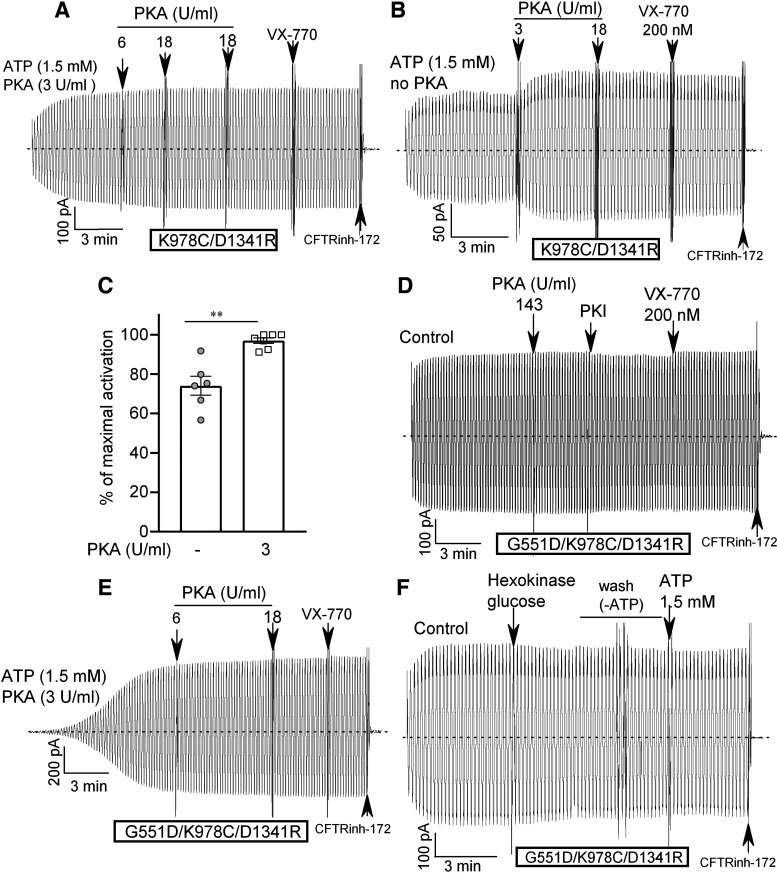
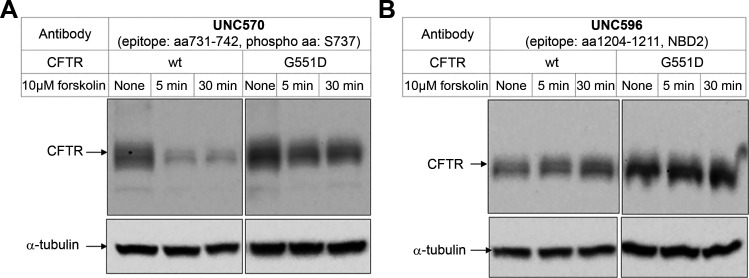
G551D is a major disease-associated gating mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) protein, an ATP- and phosphorylation-dependent chloride channel. G551D causes severe cystic fibrosis (CF) disease by disrupting ATP-dependent channel opening; however, whether G551D affects phosphorylation-dependent channel activation is unclear. Here, we use macropatch recording and Ussing chamber approaches to demonstrate that G551D impacts on phosphorylation-dependent activation of CFTR, and PKA-mediated phosphorylation regulates the interaction between the x-loop in nucleotide-binding domain 2 (NBD2) and cytosolic loop (CL) 1. We show that G551D not only disrupts ATP-dependent channel opening but also impairs phosphorylation-dependent channel activation by largely reducing PKA sensitivity consistent with the reciprocal relationship between channel opening/gating, ligand binding, and phosphorylation. Furthermore, we identified two novel GOF mutations: D1341R in the x-loop near the ATP-binding cassette signature motif in NBD2 and D173R in CL1, each of which strongly increased PKA sensitivity both in the wild-type (WT) background and when introduced into G551D-CFTR. When D1341R was combined with a second GOF mutation (e.g., K978C in CL3), we find that the double GOF mutation maximally increased G551D channel activity such that VX-770 had no further effect. We further show that a double charge-reversal mutation of D1341R/D173R-CFTR exhibited similar PKA sensitivity when compared with WT-CFTR. Together, our results suggest that charge repulsion between D173 and D1341 of WT-CFTR normally inhibits channel activation at low PKA activity by reducing PKA sensitivity, and negative allostery by the G551D is coupled to reduced PKA sensitivity of CFTR that can be restored by second GOF mutations.
Keywords: CFTR; G551D mutation; phosphorylation; x-loop.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
Similar articles
-
An electrostatic interaction at the tetrahelix bundle promotes phosphorylation-dependent cystic fibrosis transmembrane conductance regulator (CFTR) channel opening.J Biol Chem. 2014 Oct 31;289(44):30364-30378. doi: 10.1074/jbc.M114.595710. Epub 2014 Sep 4. J Biol Chem. 2014. PMID: 25190805 Free PMC article.
-
G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects.J Gen Physiol. 2007 Apr;129(4):285-98. doi: 10.1085/jgp.200609667. Epub 2007 Mar 12. J Gen Physiol. 2007. PMID: 17353351 Free PMC article.
-
Deletion of phenylalanine 508 causes attenuated phosphorylation-dependent activation of CFTR chloride channels.J Physiol. 2000 May 1;524 Pt 3(Pt 3):637-48. doi: 10.1111/j.1469-7793.2000.00637.x. J Physiol. 2000. PMID: 10790148 Free PMC article.
-
Current insights into the role of PKA phosphorylation in CFTR channel activity and the pharmacological rescue of cystic fibrosis disease-causing mutants.Cell Mol Life Sci. 2017 Jan;74(1):57-66. doi: 10.1007/s00018-016-2388-6. Epub 2016 Oct 8. Cell Mol Life Sci. 2017. PMID: 27722768 Free PMC article. Review.
-
The gating of the CFTR channel.Cell Mol Life Sci. 2017 Jan;74(1):85-92. doi: 10.1007/s00018-016-2390-z. Epub 2016 Oct 1. Cell Mol Life Sci. 2017. PMID: 27696113 Free PMC article. Review.
Cited by
-
Rescue of alveolar wall liquid secretion blocks fatal lung injury due to influenza-staphylococcal coinfection.J Clin Invest. 2023 Oct 2;133(19):e163402. doi: 10.1172/JCI163402. J Clin Invest. 2023. PMID: 37581936 Free PMC article.
-
Novel gain-of-function mutants identify a critical region within CFTR membrane-spanning domain 2 controlling cAMP-dependent and ATP-independent channel activation.Cell Mol Life Sci. 2024 Oct 7;81(1):426. doi: 10.1007/s00018-024-05431-9. Cell Mol Life Sci. 2024. PMID: 39373784 Free PMC article.
-
Role of Protein Kinase A-Mediated Phosphorylation in CFTR Channel Activity Regulation.Front Physiol. 2021 Jun 11;12:690247. doi: 10.3389/fphys.2021.690247. eCollection 2021. Front Physiol. 2021. PMID: 34211404 Free PMC article. Review.
-
Molecular mechanisms of cystic fibrosis - how mutations lead to misfunction and guide therapy.Biosci Rep. 2022 Jul 29;42(7):BSR20212006. doi: 10.1042/BSR20212006. Biosci Rep. 2022. PMID: 35707985 Free PMC article. Review.
-
Comparing ATPase activity of ATP-binding cassette subfamily C member 4, lamprey CFTR, and human CFTR using an antimony-phosphomolybdate assay.Front Pharmacol. 2024 Feb 19;15:1363456. doi: 10.3389/fphar.2024.1363456. eCollection 2024. Front Pharmacol. 2024. PMID: 38440176 Free PMC article.
References
-
- Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, Sagel SD, Hornick DB, Konstan MW, Donaldson SH, Moss RB, Pilewski JM, Rubenstein RC, Uluer AZ, Aitken ML, Freedman SD, Rose LM, Mayer-Hamblett N, Dong Q, Zha J, Stone AJ, Olson ER, Ordoñez CL, Campbell PW, Ashlock MA, Ramsey BW. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med 363: 1991–2003, 2010. doi:10.1056/NEJMoa0909825. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources