

Keratinocyte autophagy enables the activation of keratinocytes and fibroblastsand facilitates wound healing
- PMID: 32866426
- PMCID: PMC8496719
- DOI: 10.1080/15548627.2020.1816342
Keratinocyte autophagy enables the activation of keratinocytes and fibroblastsand facilitates wound healing
Abstract
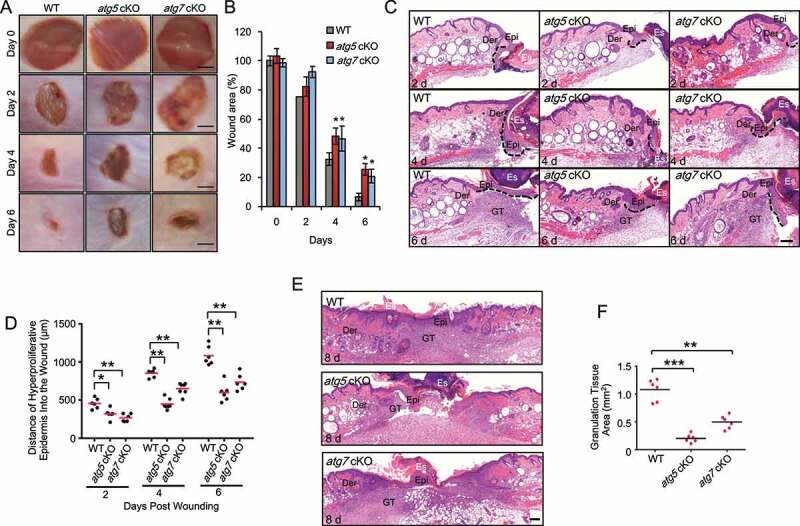
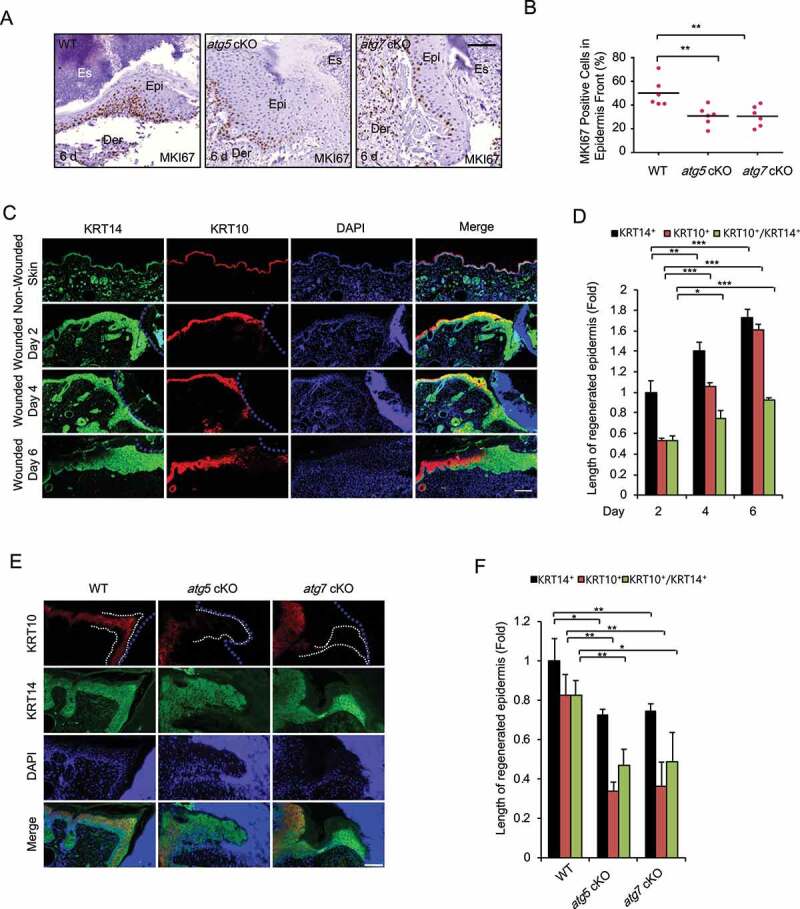
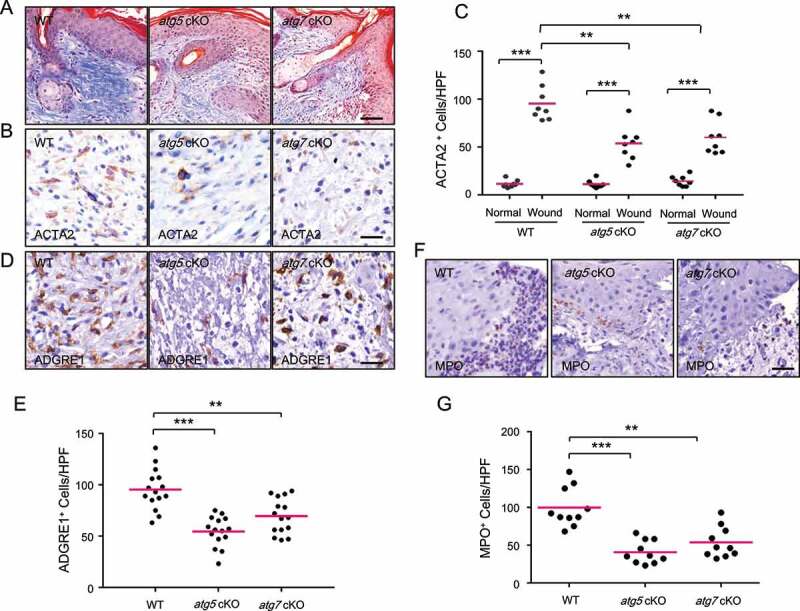
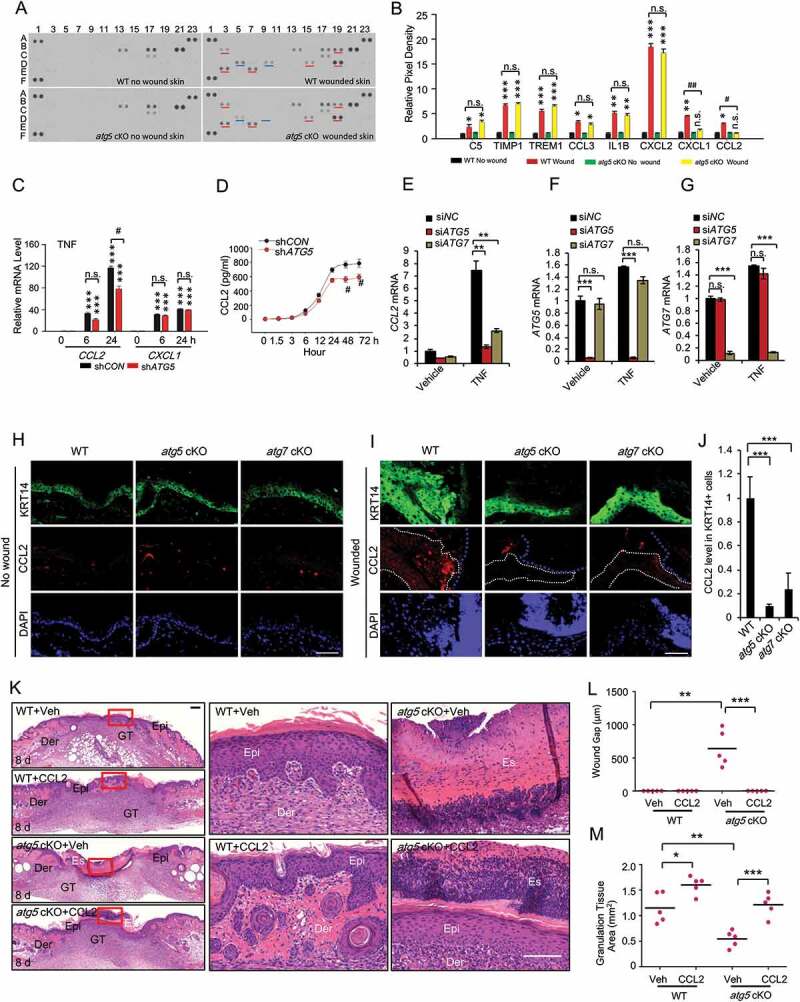
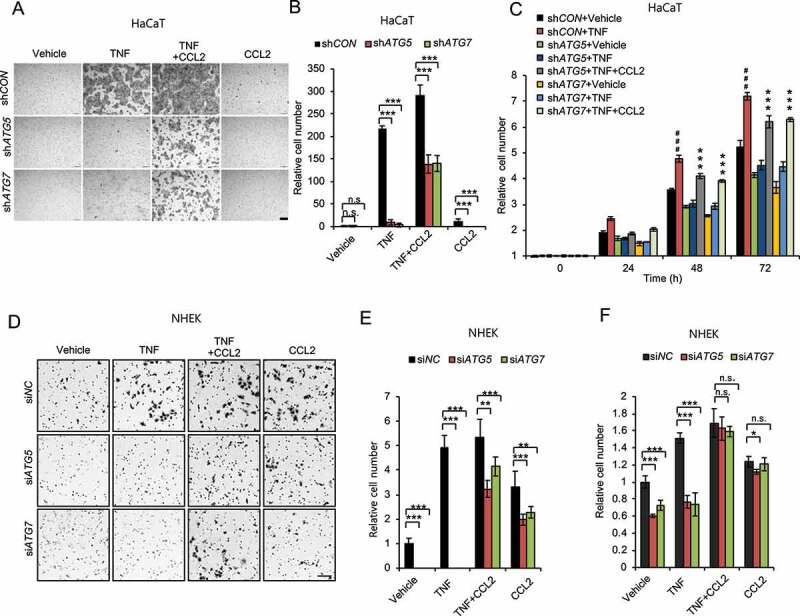
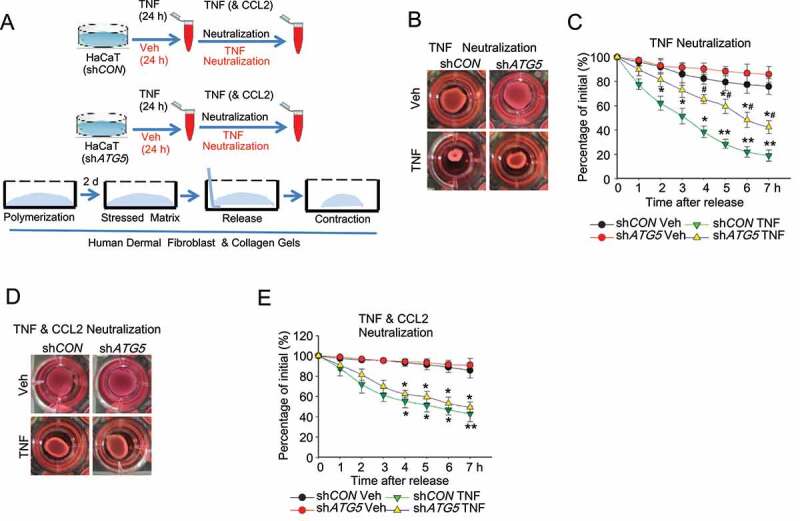
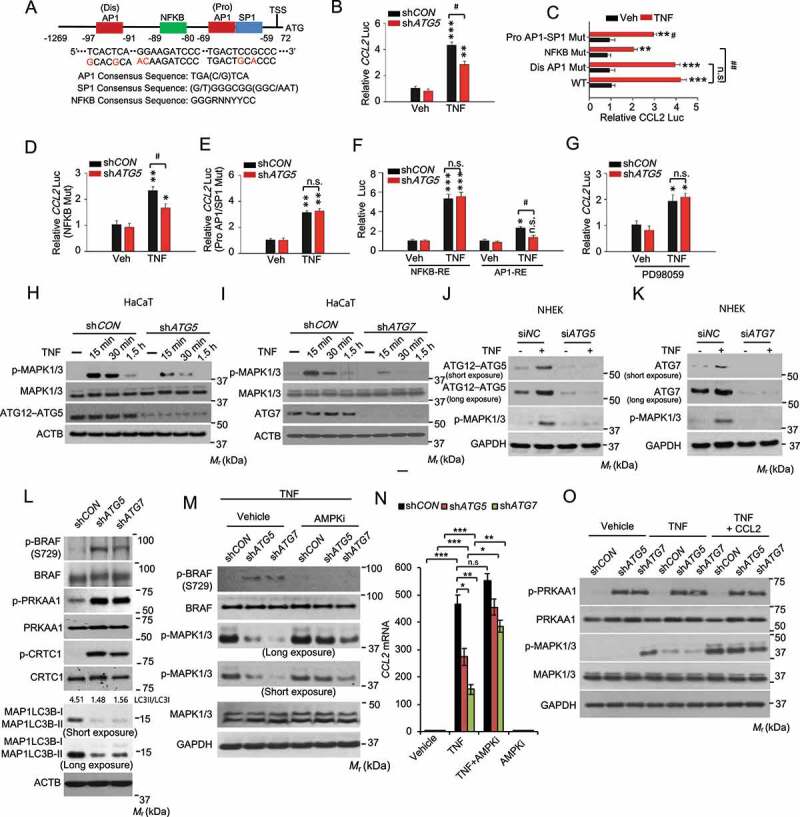
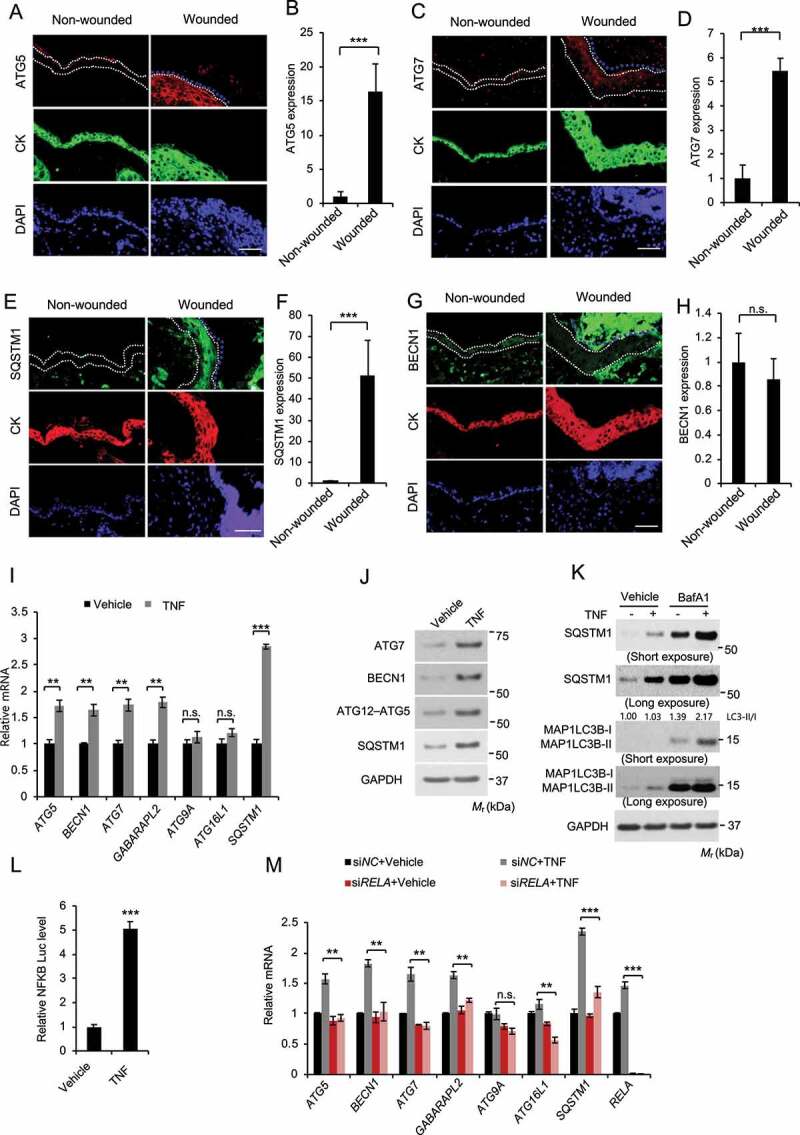
Macroautophagy/autophagy is a cellular catabolic process that is implicated in several physiological and pathological processes. However, the role of epidermal autophagy in wound healing remains unknown. Here, using mice with genetic ablation of the essential Atg5 (autophagy related 5) or Atg7 (autophagy related 7) in their epidermis to inhibit autophagy, we show that keratinocyte autophagy regulates wound healing in mice. Wounding induces the expression of autophagy genes in mouse skin. Epidermis-specific autophagy deficiency inhibits wound closure, re-epithelialization, keratinocyte proliferation and differentiation, dermal granulation tissue formation, and infiltration of immune cells including macrophages, neutrophils, and mast cells, while it does not affect angiogenesis. Using cytokine array screening, we found that autophagy deficiency inhibits the transcription and production of the cytokine CCL2/MCP-1 by TNF. At the molecular level, TNF induces autophagic flux and the expression of autophagy genes through NFKB in epidermal keratinocytes. TNF promotes CCL2 transcription through the autophagy-AMPK-BRAF-MAPK1/3/ERK-activator protein 1 (AP1) pathway. Indeed, treating mice with recombinant CCL2 can reverse the effect of autophagy deficiency in keratinocytes. At the cellular level, we found that CCL2 induction via autophagy in keratinocytes is required not only for keratinocyte migration and proliferation but also for dermal fibroblast activation. Our findings demonstrate a critical role of epidermal autophagy in wound healing in vivo and elucidate a critical molecular machinery coordinating keratinocyte-fibroblast interaction in skin repair.Abbreviations: ACTA2/α-SMA: actin alpha 2, smooth muscle; ACTB: β-actin; ADGRE1: adhesion G protein-coupled receptor E1; AMPK: AMP-activated protein kinase; AP1: activator protein 1; AP1-RE: AP1 response element; ATG: autophagy-related; ATG16L1: autophagy related 16 like 1; BECN1: beclin 1; BRAF: B-Raf proto-oncogene, serine/threonine kinase; C5: complement C5; CCL2/MCP-1: C-C motif chemokine ligand 2; CCL3: C-C motif chemokine ligand 3; CK: cytokeratin; cKO: conditional knockout; CRTC1: CREB-regulated transcription coactivator 1; CXCL1: C-X-C motif chemokine ligand 1; CXCL2: C-X-C motif chemokine ligand 2; ECM: extracellular matrix; EGF: epidermal growth factor; FGF7: fibroblast growth factor 7; GABARAPL2: GABA type A receptor associated protein like 2; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; HBEGF: heparin binding EGF like growth factor; HPRT1: hypoxanthine phosphoribosyltransferase 1; IHC: immunohistochemical; IL1B: interleukin 1 beta; KRT10: keratin 10; KRT14: keratin 14; MAP1LC3B/LC3B-I/II: microtubule-associated protein 1 light chain 3 beta; MAPK1/3/ERK: mitogen-activated protein kinase 1/3; MKI67/Ki-67: marker of proliferation; MPO: myeloperoxidase; NFKB: NF-kappa B, nuclear factor kappa-light-chain-enhancer of activated B cells; NFKB-RE: NFKB response element; PDGF: platelet-derived growth factor; PECAM1: platelet and endothelial cell adhesion molecule 1; PRKAA1: protein kinase AMP-activated catalytic subunit alpha 1; RELA/p65: RELA proto-oncogene, NFKB subunit; shCON: small hairpin negative control; siNC: negative control; siRNA: small interfering RNA; SP1: sp1 transcription factor; SQSTM1/p62: sequestosome 1; TGFA: transforming growth factor alpha; TGFB1: transforming growth factor beta 1; TIMP1: TIMP metallopeptidase inhibitor 1; TNF/TNF-alpha: tumor necrosis factor; TREM1: triggering receptor expressed on myeloid cells 1; WT: wild-type.
Keywords: Autophagy; CCL2/MCP-1; TNF; differentiation; fibroblast; inflammation; keratinocyte; migration; proliferation; wound healing.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Autophagy-based unconventional secretion of HMGB1 by keratinocytes plays a pivotal role in psoriatic skin inflammation.Autophagy. 2021 Feb;17(2):529-552. doi: 10.1080/15548627.2020.1725381. Epub 2020 Feb 16. Autophagy. 2021. PMID: 32019420 Free PMC article.
-
Autophagy regulator BECN1 suppresses mammary tumorigenesis driven by WNT1 activation and following parity.Autophagy. 2014;10(11):2036-52. doi: 10.4161/auto.34398. Epub 2014 Oct 30. Autophagy. 2014. PMID: 25483966 Free PMC article.
-
A defective lysophosphatidic acid-autophagy axis increases miscarriage risk by restricting decidual macrophage residence.Autophagy. 2022 Oct;18(10):2459-2480. doi: 10.1080/15548627.2022.2039000. Epub 2022 Feb 27. Autophagy. 2022. PMID: 35220880 Free PMC article.
-
Autophagy in the physiological endometrium and cancer.Autophagy. 2021 May;17(5):1077-1095. doi: 10.1080/15548627.2020.1752548. Epub 2020 May 13. Autophagy. 2021. PMID: 32401642 Free PMC article. Review.
-
The ménage à trois of autophagy, lipid droplets and liver disease.Autophagy. 2022 Jan;18(1):50-72. doi: 10.1080/15548627.2021.1895658. Epub 2021 Apr 2. Autophagy. 2022. PMID: 33794741 Free PMC article. Review.
Cited by
-
Improved pharmacodynamics of epidermal growth factor via microneedles-based self-powered transcutaneous electrical stimulation.Nat Commun. 2022 Nov 14;13(1):6908. doi: 10.1038/s41467-022-34716-5. Nat Commun. 2022. PMID: 36376334 Free PMC article.
-
Shining Light on Autophagy in Skin Pigmentation and Pigmentary Disorders.Cells. 2022 Sep 26;11(19):2999. doi: 10.3390/cells11192999. Cells. 2022. PMID: 36230960 Free PMC article. Review.
-
Autophagy and skin wound healing.Burns Trauma. 2022 Feb 16;10:tkac003. doi: 10.1093/burnst/tkac003. eCollection 2022. Burns Trauma. 2022. PMID: 35187180 Free PMC article. Review.
-
Compound 13 Promotes Epidermal Healing in Mouse Fetuses via Activation of AMPK.Biomedicines. 2023 Mar 27;11(4):1013. doi: 10.3390/biomedicines11041013. Biomedicines. 2023. PMID: 37189631 Free PMC article.
-
ECM-regulation of autophagy: The yin and the yang of autophagy during wound healing.Matrix Biol. 2021 Jun;100-101:197-206. doi: 10.1016/j.matbio.2020.12.006. Epub 2021 Jan 6. Matrix Biol. 2021. PMID: 33421547 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous