

Novel CAPN1 mutations extend the phenotypic heterogeneity in combined spastic paraplegia and ataxia
- PMID: 32860341
- PMCID: PMC7545613
- DOI: 10.1002/acn3.51169
Novel CAPN1 mutations extend the phenotypic heterogeneity in combined spastic paraplegia and ataxia
Abstract
Objective: Recessive mutations in the CAPN1 gene have recently been identified in spastic paraplegia 76 (SPG76), a complex hereditary spastic paraplegia (HSP) that is combined with cerebellar ataxia, resulting in an ataxia-spasticity disease spectrum. This study aims to assess the influence of CAPN1 variants on the occurrence of SPG76 and identify factors potentially contributing to phenotypic heterogeneity.
Methods: We screened a cohort of 240 unrelated HSP families for variants in CAPN1 using high-throughput sequencing analysis. We described in detail the clinical and genetic features of the SPG76 patients in our cohort and summarized all reported cases.
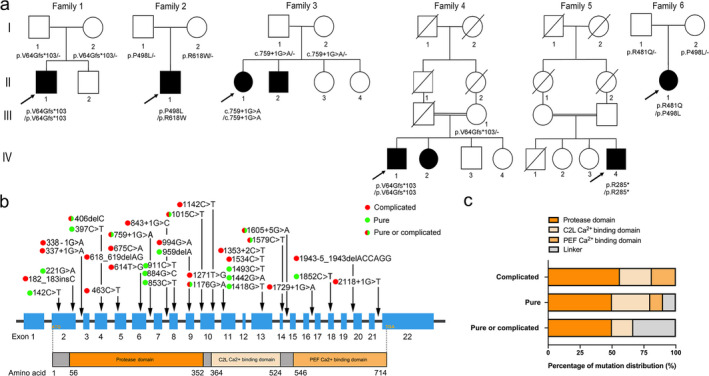
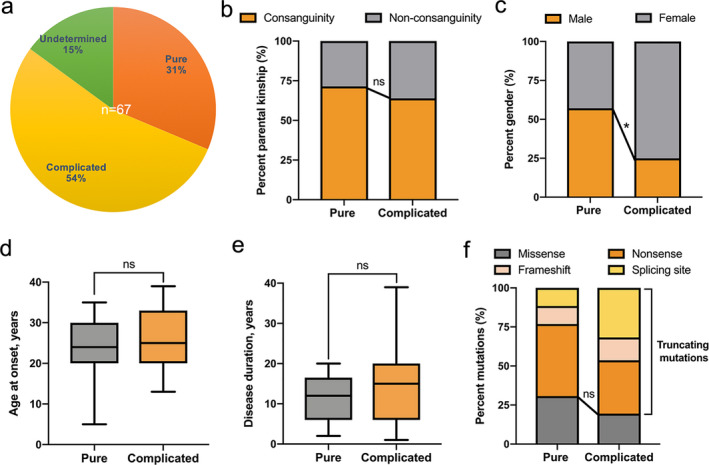
Results: Six unreported CAPN1-associated families containing eight patients with or without cerebellar ataxia were found in our cohort of HSP cases. These patients carried three previously reported homozygous truncating mutations (p.V64Gfs* 103, c.759+1G>A, and p.R285* ), and three additional novel compound heterozygous missense mutations (p.R481Q, p.P498L, and p.R618W). Lower limbs spasticity, hyperreflexia, and Babinski signs developed in about 94% of patients, with ataxia developing in 63% of cases. In total, 33 pathogenic mutations were distributed along the three reported functional domains of calpain-1 protein, encoded by CAPN1, with no hotspot region. A comparison of gender distribution between the two groups indicated that female SPG76 patients were significantly more likely to present with complicated HSP than male patients (P = 0.015).
Interpretation: Our study supports the clinically heterogeneous inter- and intra-family variability of SPG76 patients, and demonstrates that gender and calpain-1 linker structure may contribute to clinical heterogeneity in SPG76 cases.
© 2020 The Authors. Annals of Clinical and Translational Neurology published by Wiley Periodicals, Inc on behalf of American Neurological Association.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Increasing involvement of CAPN1 variants in spastic ataxias and phenotype-genotype correlations.Neurogenetics. 2021 Mar;22(1):71-79. doi: 10.1007/s10048-020-00633-2. Epub 2021 Jan 23. Neurogenetics. 2021. PMID: 33486633 Free PMC article.
-
Spastic paraplegia type 76 due to novel CAPN1 mutations: three case reports with literature review.Neurogenetics. 2023 Oct;24(4):243-250. doi: 10.1007/s10048-023-00726-8. Epub 2023 Jul 19. Neurogenetics. 2023. PMID: 37468791 Review.
-
Two novel homozygous mutations of CAPN1 in Chinese patients with hereditary spastic paraplegia and literatures review.Orphanet J Rare Dis. 2019 Apr 25;14(1):83. doi: 10.1186/s13023-019-1053-1. Orphanet J Rare Dis. 2019. PMID: 31023339 Free PMC article. Review.
-
CAPN1 mutations: Expanding the CAPN1-related phenotype: From hereditary spastic paraparesis to spastic ataxia.Eur J Med Genet. 2019 Dec;62(12):103605. doi: 10.1016/j.ejmg.2018.12.010. Epub 2018 Dec 17. Eur J Med Genet. 2019. PMID: 30572172
-
CAPN1 and hereditary spastic paraplegia: a novel variant in an Iranian family and overview of the genotype-phenotype correlation.Int J Neurosci. 2021 Oct;131(10):962-974. doi: 10.1080/00207454.2020.1763344. Epub 2020 May 13. Int J Neurosci. 2021. PMID: 32352326
Cited by
-
Hereditary spastic paraplegia: Novel insights into the pathogenesis and management.SAGE Open Med. 2023 Dec 29;12:20503121231221941. doi: 10.1177/20503121231221941. eCollection 2024. SAGE Open Med. 2023. PMID: 38162912 Free PMC article. Review.
-
Increasing involvement of CAPN1 variants in spastic ataxias and phenotype-genotype correlations.Neurogenetics. 2021 Mar;22(1):71-79. doi: 10.1007/s10048-020-00633-2. Epub 2021 Jan 23. Neurogenetics. 2021. PMID: 33486633 Free PMC article.
-
Spastic paraplegia type 76 due to novel CAPN1 mutations: three case reports with literature review.Neurogenetics. 2023 Oct;24(4):243-250. doi: 10.1007/s10048-023-00726-8. Epub 2023 Jul 19. Neurogenetics. 2023. PMID: 37468791 Review.
-
PLP1 gene mutations cause spastic paraplegia type 2 in three families.Ann Clin Transl Neurol. 2023 Mar;10(3):328-338. doi: 10.1002/acn3.51722. Epub 2023 Jan 9. Ann Clin Transl Neurol. 2023. PMID: 36622199 Free PMC article.
-
A Compound Heterozygous Mutation in Calpain 1 Identifies a New Genetic Cause for Spinal Muscular Atrophy Type 4 (SMA4).Front Genet. 2022 Jan 19;12:801253. doi: 10.3389/fgene.2021.801253. eCollection 2021. Front Genet. 2022. PMID: 35126465 Free PMC article.
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
