

Cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of TMEM16A in vivo
- PMID: 32859916
- PMCID: PMC7455562
- DOI: 10.1038/s41467-020-18104-5
Cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of TMEM16A in vivo
Abstract
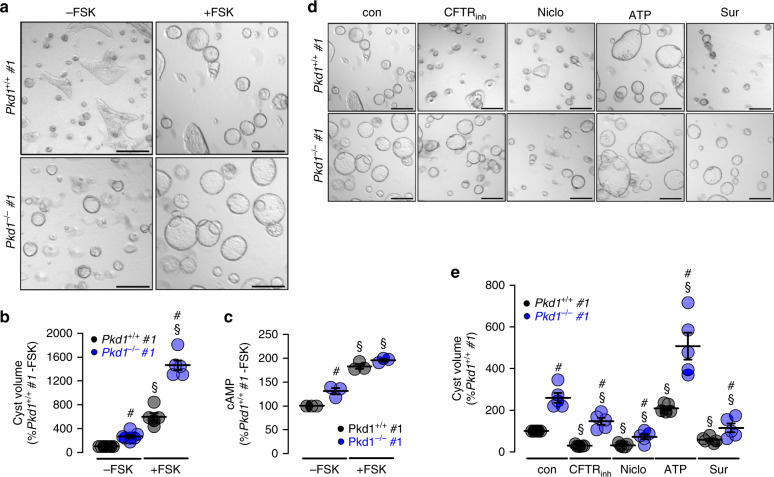
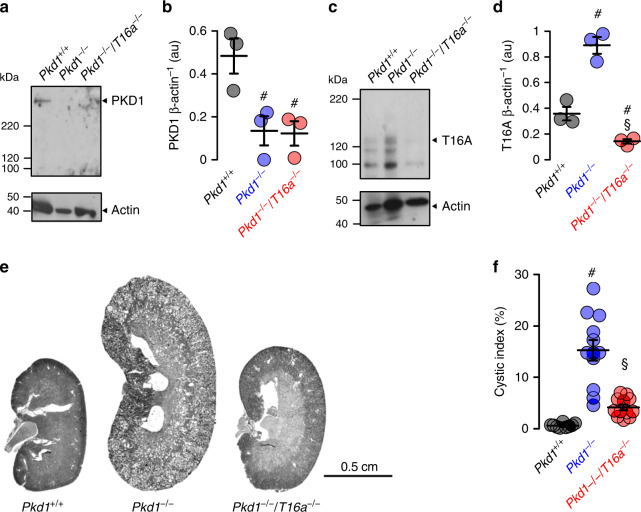
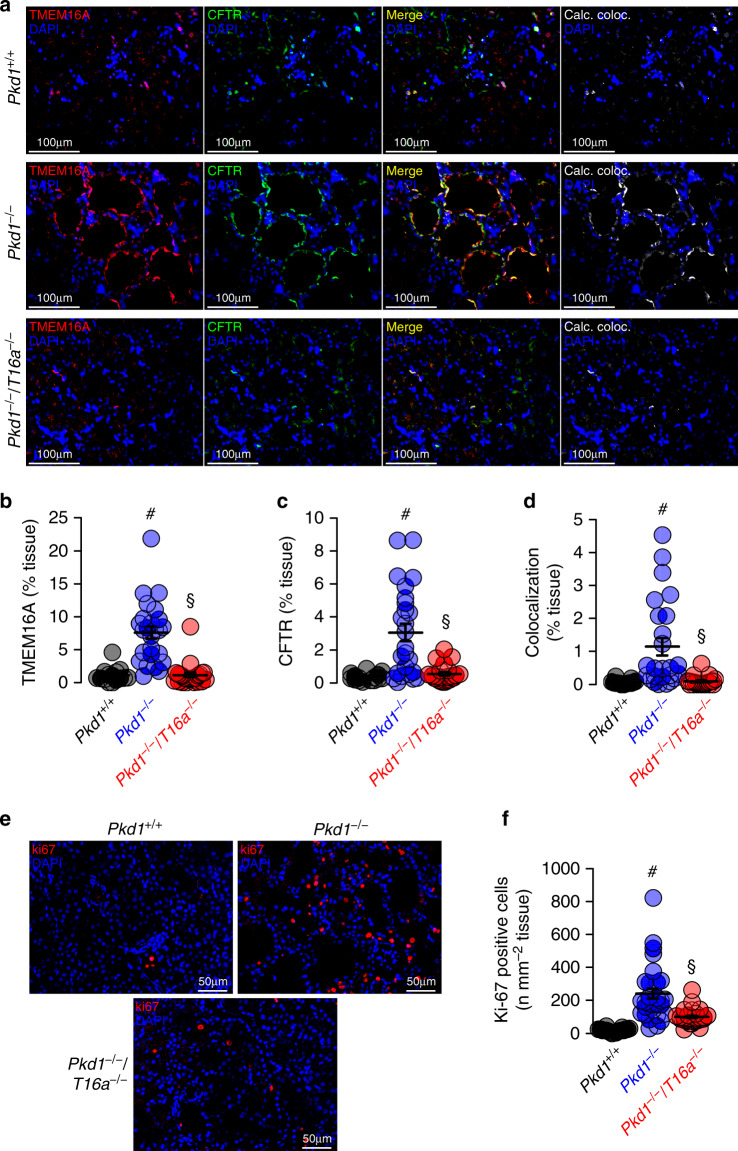
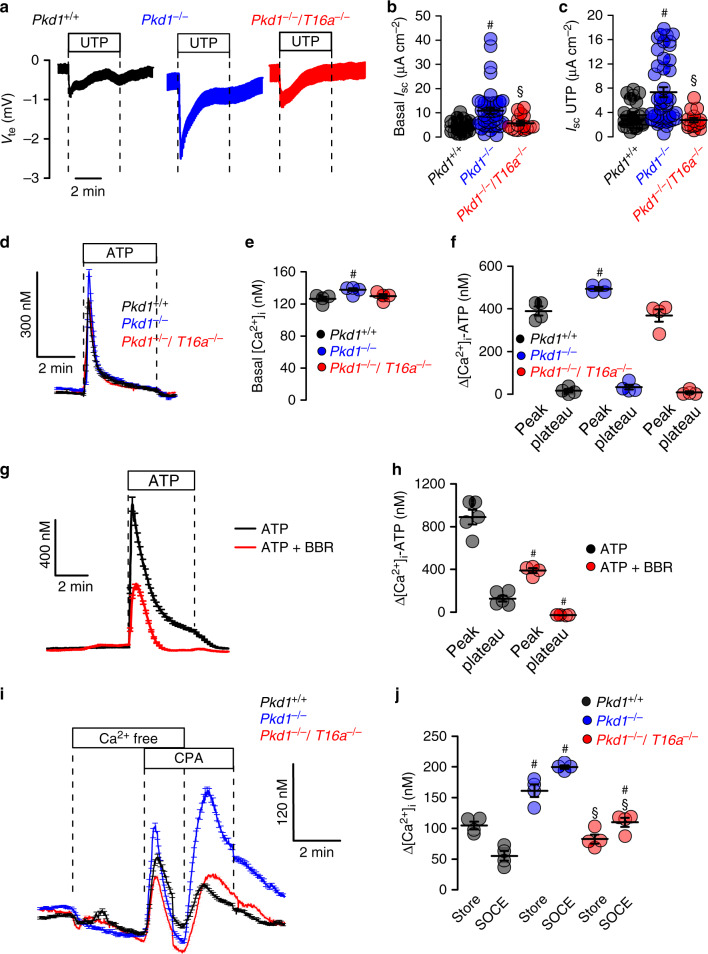
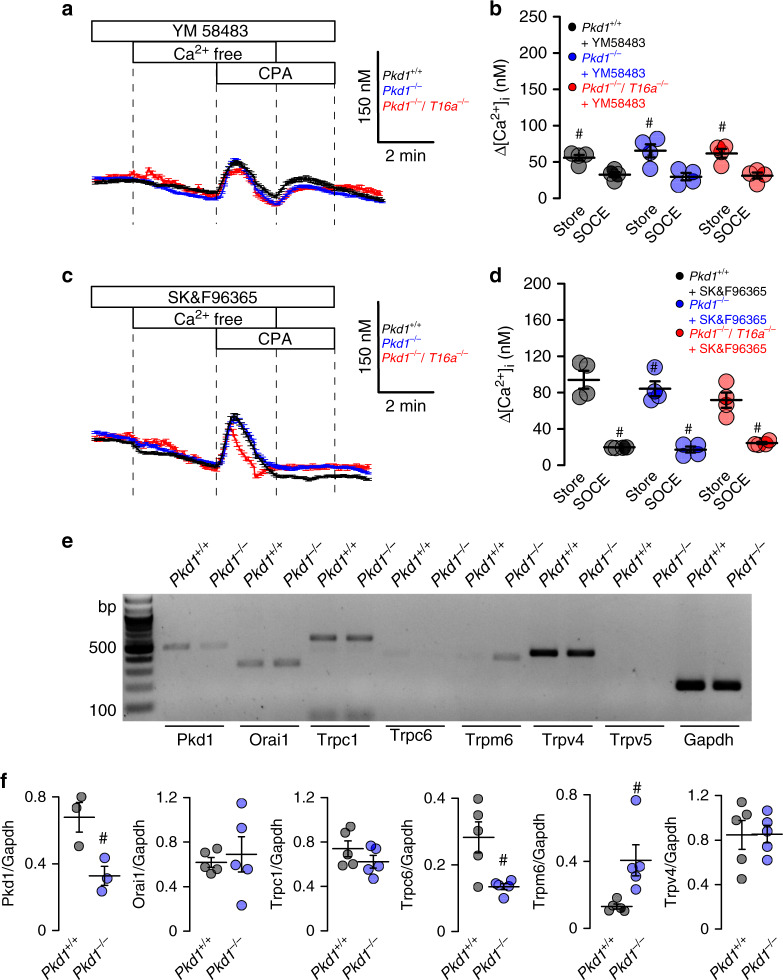
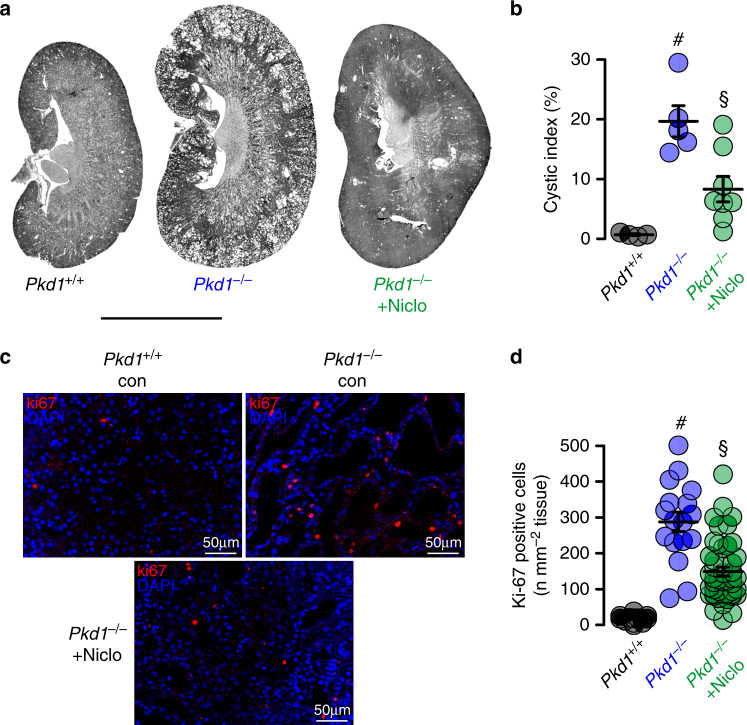
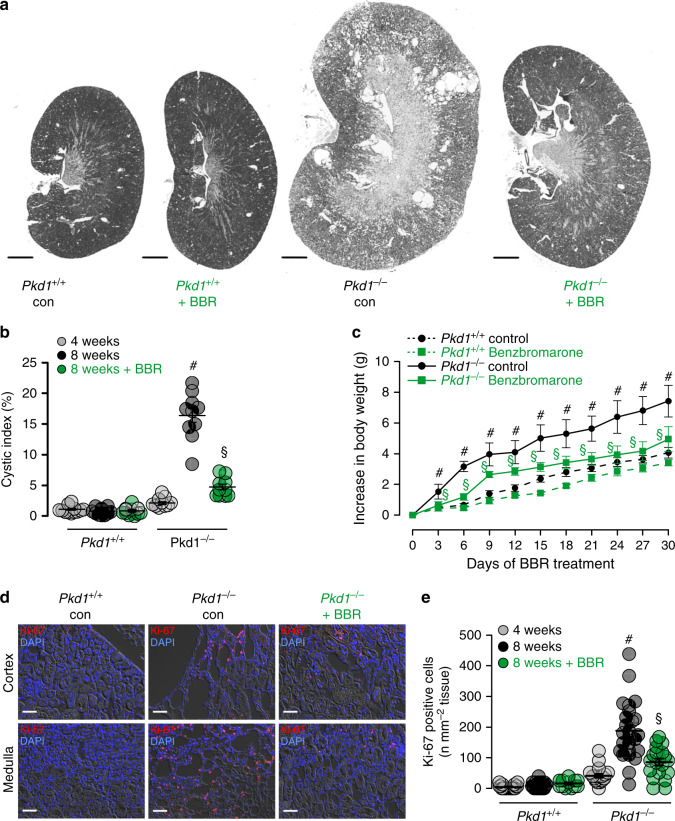
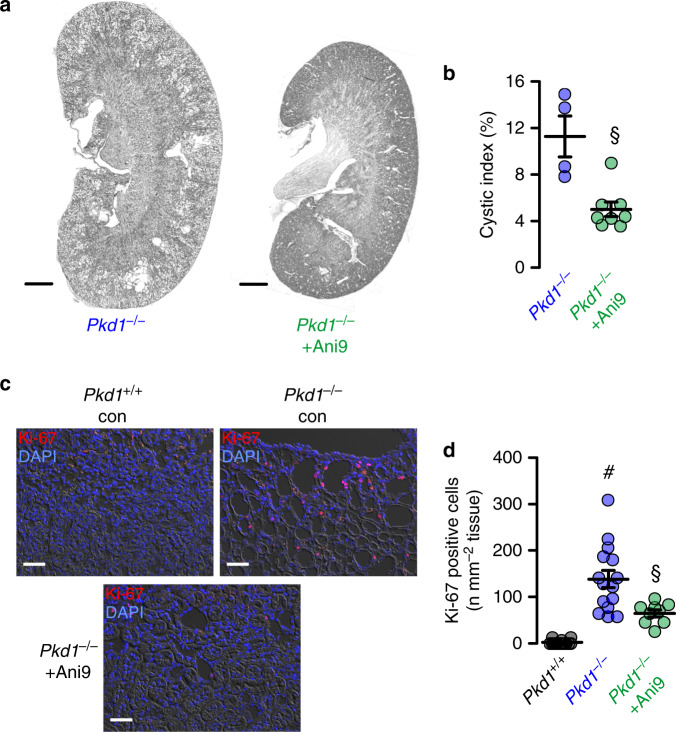
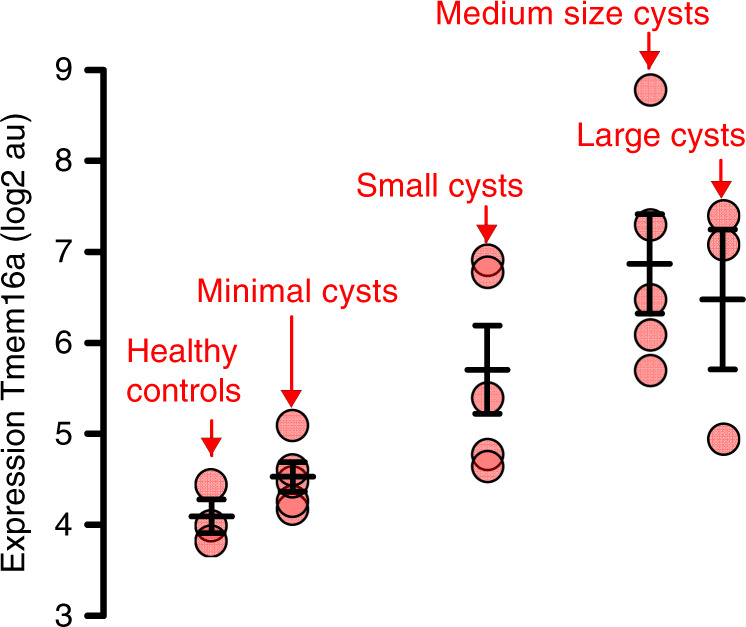
In autosomal dominant polycystic kidney disease (ADPKD) multiple bilateral renal cysts gradually enlarge, leading to a decline in renal function. Transepithelial chloride secretion through cystic fibrosis transmembrane conductance regulator (CFTR) and TMEM16A (anoctamin 1) are known to drive cyst enlargement. Here we demonstrate that loss of Pkd1 increased expression of TMEM16A and CFTR and Cl- secretion in murine kidneys, with TMEM16A essentially contributing to cyst growth. Upregulated TMEM16A enhanced intracellular Ca2+ signaling and proliferation of Pkd1-deficient renal epithelial cells. In contrast, increase in Ca2+ signaling, cell proliferation and CFTR expression was not observed in Pkd1/Tmem16a double knockout mice. Knockout of Tmem16a or inhibition of TMEM16A in vivo by the FDA-approved drugs niclosamide and benzbromarone, as well as the TMEM16A-specific inhibitor Ani9 largely reduced cyst enlargement and abnormal cyst cell proliferation. The present data establish a therapeutic concept for the treatment of ADPKD.
Conflict of interest statement
The authors declare no competing interests
Figures
Similar articles
-
The chloride channel CFTR is not required for cyst growth in an ADPKD mouse model.FASEB J. 2021 Oct;35(10):e21897. doi: 10.1096/fj.202100843R. FASEB J. 2021. PMID: 34473378
-
TMEM16A drives renal cyst growth by augmenting Ca2+ signaling in M1 cells.J Mol Med (Berl). 2020 May;98(5):659-671. doi: 10.1007/s00109-020-01894-y. Epub 2020 Mar 18. J Mol Med (Berl). 2020. PMID: 32185407 Free PMC article.
-
Gender-Dependent Phenotype in Polycystic Kidney Disease Is Determined by Differential Intracellular Ca2+ Signals.Int J Mol Sci. 2021 Jun 2;22(11):6019. doi: 10.3390/ijms22116019. Int J Mol Sci. 2021. PMID: 34199520 Free PMC article.
-
[Inhibitors of intra-cystic secretion: novel therapies in ADPKD (Autosomal Dominant Polycystic Kidney Disease)].G Ital Nefrol. 2013 Jan-Feb;30(1):gin/30.1.2. G Ital Nefrol. 2013. PMID: 23832438 Review. Italian.
-
Cell Proliferation and Apoptosis in ADPKD.Adv Exp Med Biol. 2016;933:25-34. doi: 10.1007/978-981-10-2041-4_3. Adv Exp Med Biol. 2016. PMID: 27730432 Review.
Cited by
-
Scalable production of uniform and mature organoids in a 3D geometrically-engineered permeable membrane.Nat Commun. 2024 Oct 31;15(1):9420. doi: 10.1038/s41467-024-53073-z. Nat Commun. 2024. PMID: 39482314 Free PMC article.
-
Impact of Pals1 on Expression and Localization of Transporters Belonging to the Solute Carrier Family.Front Mol Biosci. 2022 Feb 16;9:792829. doi: 10.3389/fmolb.2022.792829. eCollection 2022. Front Mol Biosci. 2022. PMID: 35252349 Free PMC article.
-
Abnormal Iron and Lipid Metabolism Mediated Ferroptosis in Kidney Diseases and Its Therapeutic Potential.Metabolites. 2022 Jan 10;12(1):58. doi: 10.3390/metabo12010058. Metabolites. 2022. PMID: 35050181 Free PMC article. Review.
-
CLCA1 Regulates Airway Mucus Production and Ion Secretion Through TMEM16A.Int J Mol Sci. 2021 May 12;22(10):5133. doi: 10.3390/ijms22105133. Int J Mol Sci. 2021. PMID: 34066250 Free PMC article.
-
The Controversial Role of Fibrosis in Autosomal Dominant Polycystic Kidney Disease.Int J Mol Sci. 2020 Nov 25;21(23):8936. doi: 10.3390/ijms21238936. Int J Mol Sci. 2020. PMID: 33255651 Free PMC article. Review.
References
-
- Nauli SM, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003;33:129–137. - PubMed
-
- Kunzelmann K. The cystic fibrosis transmembrane conductance regulator and its function in epithelial transport. Rev. Physiol. Biochem. Pharmacol. 1999;137:1–70. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
