

Development of Charge-Augmented Three-Point Water Model (CAIPi3P) for Accurate Simulations of Intrinsically Disordered Proteins
- PMID: 32859072
- PMCID: PMC7504337
- DOI: 10.3390/ijms21176166
Development of Charge-Augmented Three-Point Water Model (CAIPi3P) for Accurate Simulations of Intrinsically Disordered Proteins
Abstract
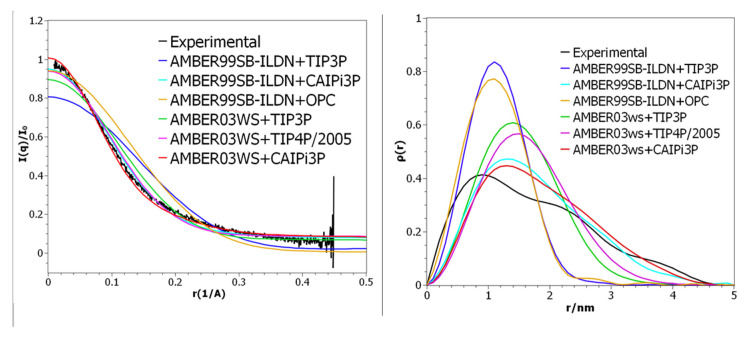
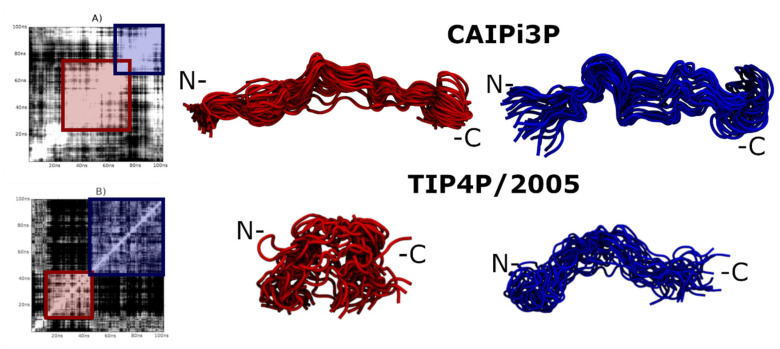
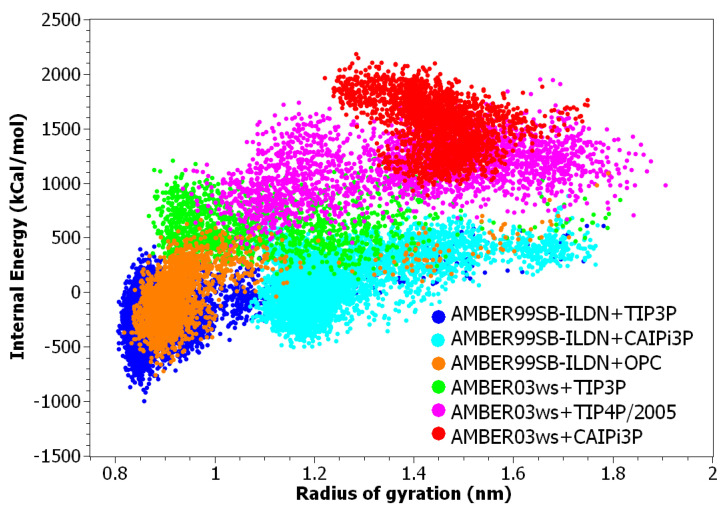
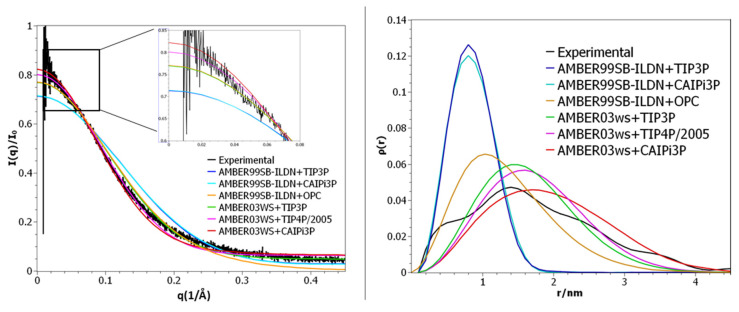
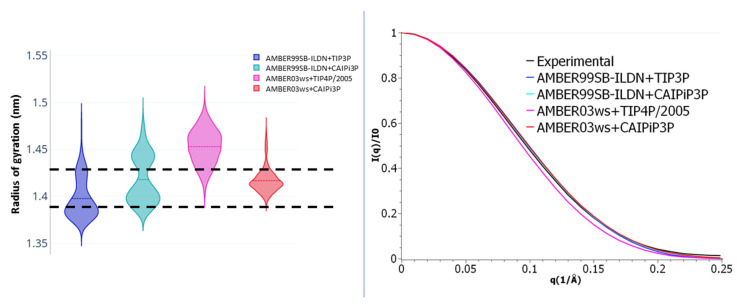
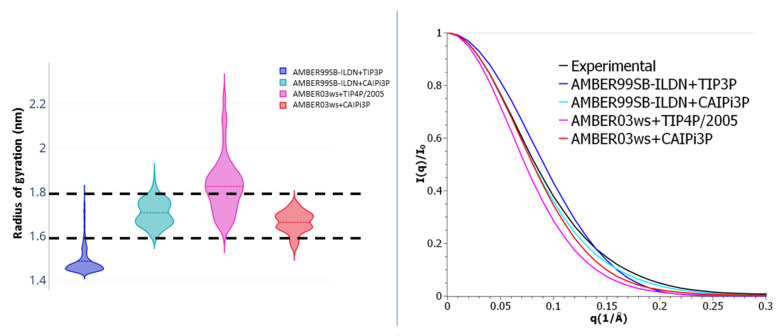
Intrinsically disordered proteins (IDPs) are molecules without a fixed tertiary structure, exerting crucial roles in cellular signalling, growth and molecular recognition events. Due to their high plasticity, IDPs are very challenging in experimental and computational structural studies. To provide detailed atomic insight in IDPs' dynamics governing their functional mechanisms, all-atom molecular dynamics (MD) simulations are widely employed. However, the current generalist force fields and solvent models are unable to generate satisfactory ensembles for IDPs when compared to existing experimental data. In this work, we present a new solvation model, denoted as the Charge-Augmented Three-Point Water Model for Intrinsically Disordered Proteins (CAIPi3P). CAIPi3P has been generated by performing a systematic scan of atomic partial charges assigned to the widely popular molecular scaffold of the three-point TIP3P water model. We found that explicit solvent MD simulations employing CAIPi3P solvation considerably improved the small-angle X-ray scattering (SAXS) scattering profiles for three different IDPs. Not surprisingly, this improvement was further enhanced by using CAIPi3P water in combination with the protein force field parametrized for IDPs. We also demonstrated the applicability of CAIPi3P to molecular systems containing structured as well as intrinsically disordered regions/domains. Our results highlight the crucial importance of solvent effects for generating molecular ensembles of IDPs which reproduce the experimental data available. Hence, we conclude that our newly developed CAIPi3P solvation model is a valuable tool for molecular simulations of intrinsically disordered proteins and assessing their molecular dynamics.
Keywords: CAIPi3P; IDPs; intrinsically disordered proteins; molecular dynamics; water models.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
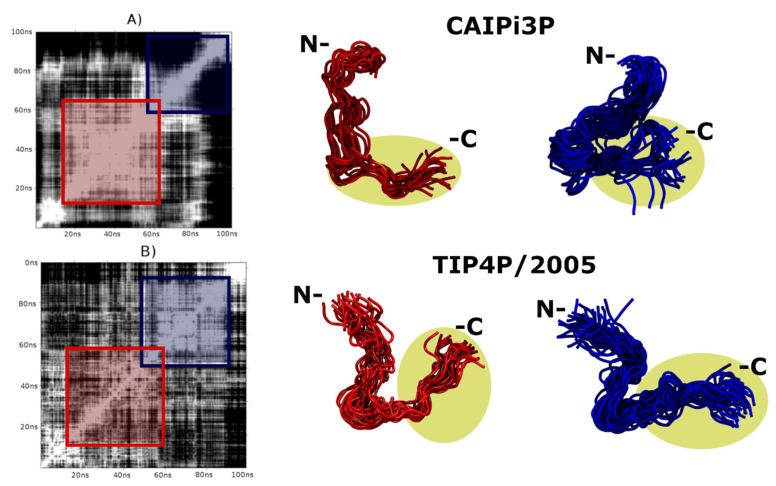
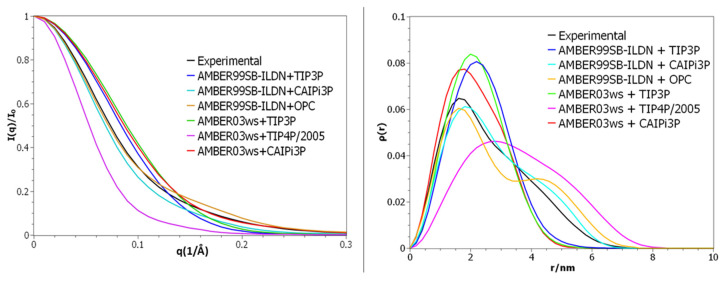
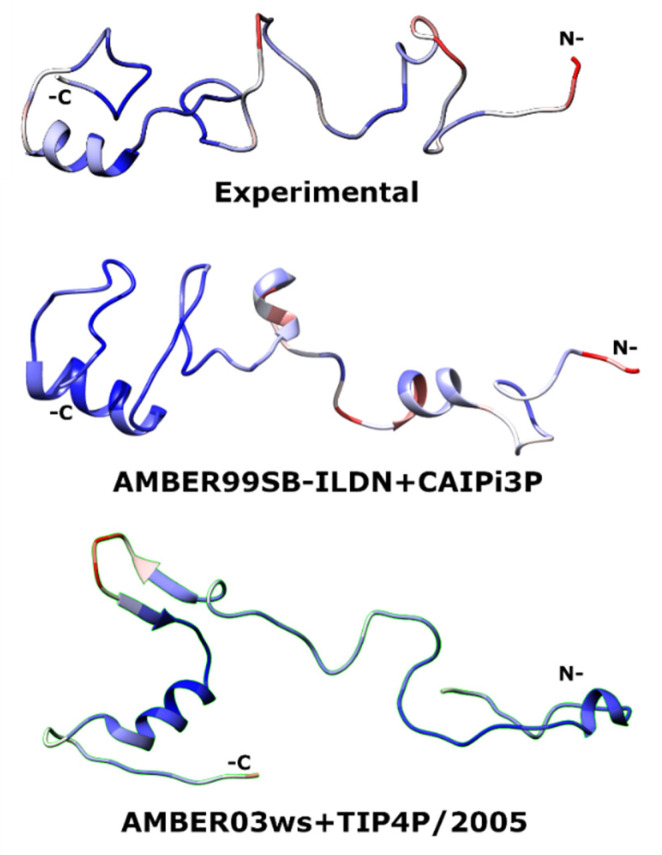
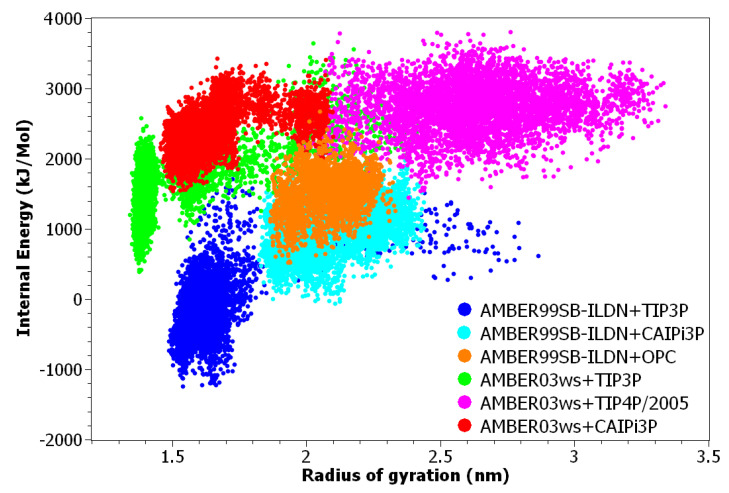
Similar articles
-
Molecular Dynamics Simulations Combined with Nuclear Magnetic Resonance and/or Small-Angle X-ray Scattering Data for Characterizing Intrinsically Disordered Protein Conformational Ensembles.J Chem Inf Model. 2019 May 28;59(5):1743-1758. doi: 10.1021/acs.jcim.8b00928. Epub 2019 Mar 18. J Chem Inf Model. 2019. PMID: 30840442 Review.
-
Hierarchical Ensembles of Intrinsically Disordered Proteins at Atomic Resolution in Molecular Dynamics Simulations.J Chem Theory Comput. 2020 Jan 14;16(1):725-737. doi: 10.1021/acs.jctc.9b00809. Epub 2019 Dec 26. J Chem Theory Comput. 2020. PMID: 31809054
-
Intrinsically disordered protein-specific force field CHARMM36IDPSFF.Chem Biol Drug Des. 2018 Oct;92(4):1722-1735. doi: 10.1111/cbdd.13342. Epub 2018 Jul 1. Chem Biol Drug Des. 2018. PMID: 29808548
-
SAXS-Restrained Ensemble Simulations of Intrinsically Disordered Proteins with Commitment to the Principle of Maximum Entropy.J Chem Theory Comput. 2019 Sep 10;15(9):5103-5115. doi: 10.1021/acs.jctc.9b00338. Epub 2019 Aug 26. J Chem Theory Comput. 2019. PMID: 31402649
-
Force field development and simulations of intrinsically disordered proteins.Curr Opin Struct Biol. 2018 Feb;48:40-48. doi: 10.1016/j.sbi.2017.10.008. Epub 2017 Nov 5. Curr Opin Struct Biol. 2018. PMID: 29080468 Free PMC article. Review.
Cited by
-
Self-Diffusive Properties of the Intrinsically Disordered Protein Histatin 5 and the Impact of Crowding Thereon: A Combined Neutron Spectroscopy and Molecular Dynamics Simulation Study.J Phys Chem B. 2022 Feb 3;126(4):789-801. doi: 10.1021/acs.jpcb.1c08976. Epub 2022 Jan 19. J Phys Chem B. 2022. PMID: 35044776 Free PMC article.
-
Myofilament-associated proteins with intrinsic disorder (MAPIDs) and their resolution by computational modeling.Q Rev Biophys. 2023 Jan 11;56:e2. doi: 10.1017/S003358352300001X. Q Rev Biophys. 2023. PMID: 36628457 Free PMC article. Review.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
