

Endometrial Cancer as a Metabolic Disease with Dysregulated PI3K Signaling: Shedding Light on Novel Therapeutic Strategies
- PMID: 32842547
- PMCID: PMC7504460
- DOI: 10.3390/ijms21176073
Endometrial Cancer as a Metabolic Disease with Dysregulated PI3K Signaling: Shedding Light on Novel Therapeutic Strategies
Abstract
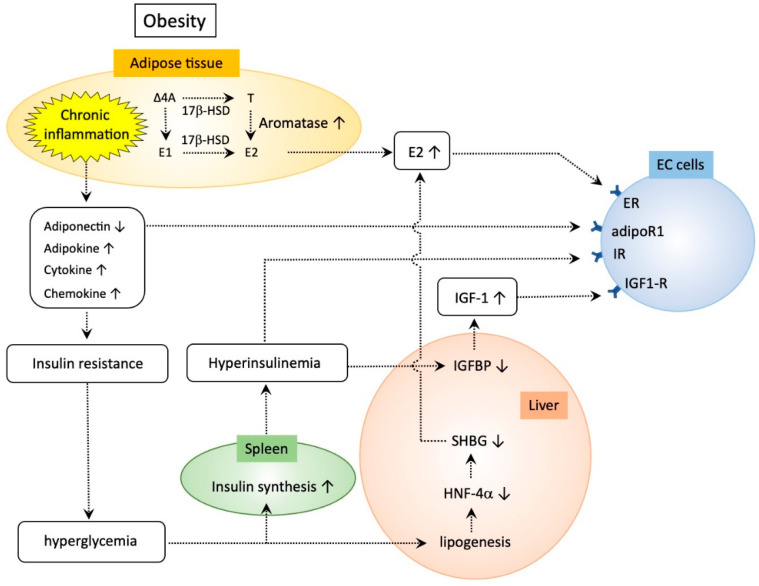
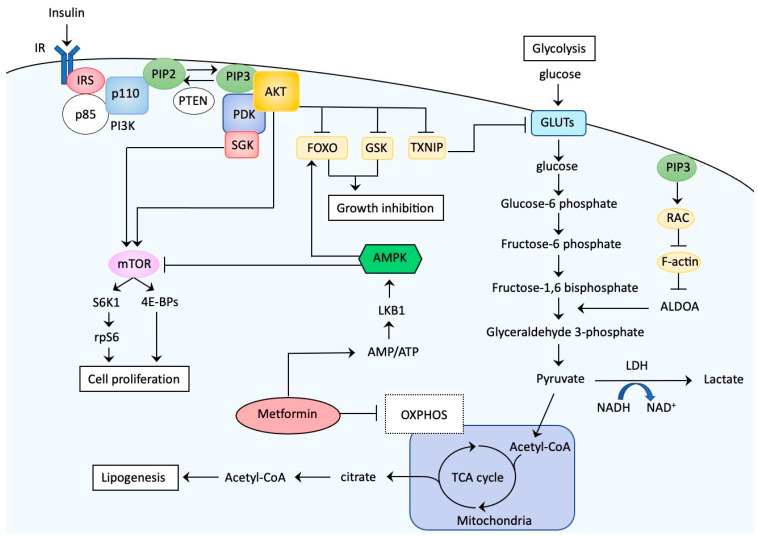
Endometrial cancer (EC) is one of the most common malignancies of the female reproductive organs. The most characteristic feature of EC is the frequent association with metabolic disorders. However, the components of these disorders that are involved in carcinogenesis remain unclear. Accumulating epidemiological studies have clearly revealed that hyperinsulinemia, which accompanies these disorders, plays central roles in the development of EC via the insulin-phosphoinositide 3 kinase (PI3K) signaling pathway as a metabolic driver. Recent comprehensive genomic analyses showed that over 90% of ECs have genomic alterations in this pathway, resulting in enhanced insulin signaling and production of optimal tumor microenvironments (TMEs). Targeting PI3K signaling is therefore an attractive treatment strategy. Several clinical trials for recurrent or advanced ECs have been attempted using PI3K-serine/threonine kinase (AKT) inhibitors. However, these agents exhibited far lower efficacy than expected, possibly due to activation of alternative pathways that compensate for the PIK3-AKT pathway and allow tumor growth, or due to adaptive mechanisms including the insulin feedback pathway that limits the efficacy of agents. Overcoming these responses with careful management of insulin levels is key to successful treatment. Further interest in specific TMEs via the insulin PI3K-pathway in obese women will provide insight into not only novel therapeutic strategies but also preventive strategies against EC.
Keywords: PI3K-AKT; endometrial cancer; hyperglycemia; hyperinsulinemia; metabolic syndrome; tumor microenvironment.
Conflict of interest statement
The author declares no conflict of interest.
Figures
Similar articles
-
Changes in protein expression due to metformin treatment and hyperinsulinemia in a human endometrial cancer cell line.PLoS One. 2021 Mar 9;16(3):e0248103. doi: 10.1371/journal.pone.0248103. eCollection 2021. PLoS One. 2021. PMID: 33690729 Free PMC article.
-
Activation of PI3K/Akt/mTOR pathway and dual inhibitors of PI3K and mTOR in endometrial cancer.Curr Med Chem. 2014;21(26):3070-80. doi: 10.2174/0929867321666140414095605. Curr Med Chem. 2014. PMID: 24735369 Review.
-
Inhibition of midkine by metformin can contribute to its anticancer effects in malignancies: A proposal mechanism of action of metformin in context of endometrial cancer prevention and therapy.Med Hypotheses. 2020 Jan;134:109420. doi: 10.1016/j.mehy.2019.109420. Epub 2019 Oct 3. Med Hypotheses. 2020. PMID: 31634770
-
Investigational PI3K/AKT/mTOR inhibitors in development for endometrial cancer.Expert Opin Investig Drugs. 2019 Feb;28(2):131-142. doi: 10.1080/13543784.2018.1558202. Epub 2018 Dec 21. Expert Opin Investig Drugs. 2019. PMID: 30574817 Review.
-
Assessing the efficacy of targeting the phosphatidylinositol 3-kinase/AKT/mTOR signaling pathway in endometrial cancer.Gynecol Oncol. 2014 May;133(2):346-52. doi: 10.1016/j.ygyno.2014.02.022. Epub 2014 Feb 19. Gynecol Oncol. 2014. PMID: 24561032
Cited by
-
Effect of integrin α7 on cell proliferation, invasion, apoptosis and the PI3K/AKT pathway, and its association with clinicopathological features in endometrial cancer.Oncol Lett. 2022 Nov 23;25(1):26. doi: 10.3892/ol.2022.13612. eCollection 2023 Jan. Oncol Lett. 2022. PMID: 36478908 Free PMC article.
-
Progestin Resistance and Corresponding Management of Abnormal Endometrial Hyperplasia and Endometrial Carcinoma.Cancers (Basel). 2022 Dec 15;14(24):6210. doi: 10.3390/cancers14246210. Cancers (Basel). 2022. PMID: 36551694 Free PMC article. Review.
-
Fertility-Sparing Treatment for Endometrial Cancer: Oncological and Obstetric Outcomes in Combined Therapies with Levonorgestrel Intrauterine Device.Cancers (Basel). 2022 Apr 26;14(9):2170. doi: 10.3390/cancers14092170. Cancers (Basel). 2022. PMID: 35565299 Free PMC article.
-
Grading of endometrial cancer using 1H HR-MAS NMR-based metabolomics.Sci Rep. 2021 Sep 13;11(1):18160. doi: 10.1038/s41598-021-97505-y. Sci Rep. 2021. PMID: 34518615 Free PMC article.
-
The way to precision medicine in gynecologic cancers: The first case report of an exceptional response to alpelisib in a PIK3CA-mutated endometrial cancer.Front Oncol. 2023 Jan 13;12:1088962. doi: 10.3389/fonc.2022.1088962. eCollection 2022. Front Oncol. 2023. PMID: 36713525 Free PMC article.
References
-
- Cancer Information Service . Cancer Registry and Statistics (Vital Statistics of Japan) Cancer Information Service; National Cancer Center, Tokyo, Japan: 2016.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
