

Systematic determination of the mitochondrial proportion in human and mice tissues for single-cell RNA-sequencing data quality control
- PMID: 32840568
- PMCID: PMC8599307
- DOI: 10.1093/bioinformatics/btaa751
Systematic determination of the mitochondrial proportion in human and mice tissues for single-cell RNA-sequencing data quality control
Abstract
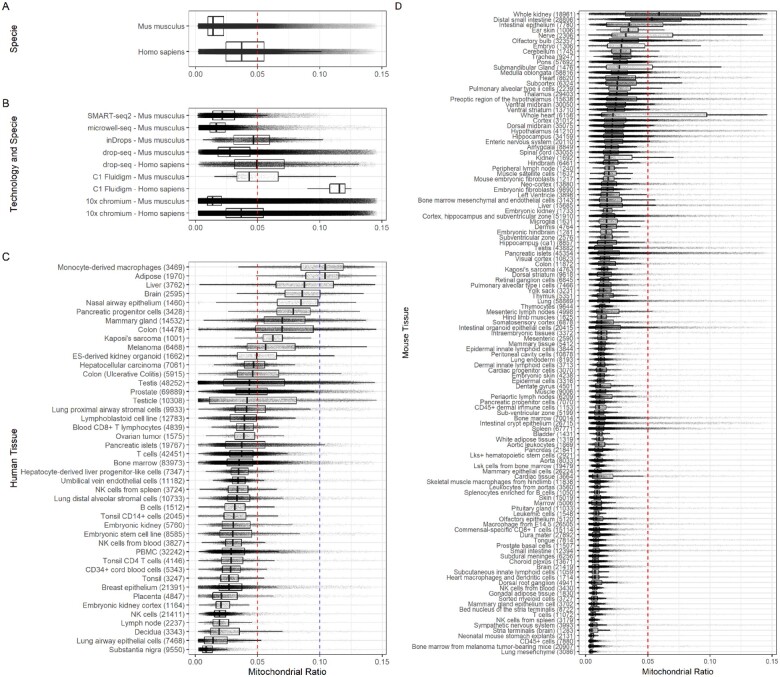
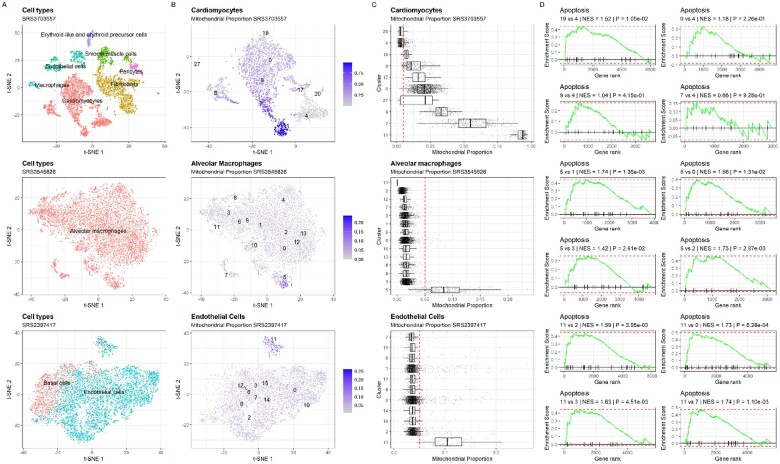
Motivation: Quality control (QC) is a critical step in single-cell RNA-seq (scRNA-seq) data analysis. Low-quality cells are removed from the analysis during the QC process to avoid misinterpretation of the data. An important QC metric is the mitochondrial proportion (mtDNA%), which is used as a threshold to filter out low-quality cells. Early publications in the field established a threshold of 5% and since then, it has been used as a default in several software packages for scRNA-seq data analysis, and adopted as a standard in many scRNA-seq studies. However, the validity of using a uniform threshold across different species, single-cell technologies, tissues and cell types has not been adequately assessed.
Results: We systematically analyzed 5 530 106 cells reported in 1349 annotated datasets available in the PanglaoDB database and found that the average mtDNA% in scRNA-seq data across human tissues is significantly higher than in mouse tissues. This difference is not confounded by the platform used to generate the data. Based on this finding, we propose new reference values of the mtDNA% for 121 tissues of mouse and 44 tissues of humans. In general, for mouse tissues, the 5% threshold performs well to distinguish between healthy and low-quality cells. However, for human tissues, the 5% threshold should be reconsidered as it fails to accurately discriminate between healthy and low-quality cells in 29.5% (13 of 44) tissues analyzed. We conclude that omitting the mtDNA% QC filter or adopting a suboptimal mtDNA% threshold may lead to erroneous biological interpretations of scRNA-seq data.
Availabilityand implementation: The code used to download datasets, perform the analyzes and produce the figures is available at https://github.com/dosorio/mtProportion.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2020. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
Similar articles
-
scQCEA: a framework for annotation and quality control report of single-cell RNA-sequencing data.BMC Genomics. 2023 Jul 6;24(1):381. doi: 10.1186/s12864-023-09447-6. BMC Genomics. 2023. PMID: 37415108 Free PMC article.
-
miQC: An adaptive probabilistic framework for quality control of single-cell RNA-sequencing data.PLoS Comput Biol. 2021 Aug 24;17(8):e1009290. doi: 10.1371/journal.pcbi.1009290. eCollection 2021 Aug. PLoS Comput Biol. 2021. PMID: 34428202 Free PMC article.
-
Cell-level somatic mutation detection from single-cell RNA sequencing.Bioinformatics. 2019 Nov 1;35(22):4679-4687. doi: 10.1093/bioinformatics/btz288. Bioinformatics. 2019. PMID: 31028395 Free PMC article.
-
Machine learning and statistical methods for clustering single-cell RNA-sequencing data.Brief Bioinform. 2020 Jul 15;21(4):1209-1223. doi: 10.1093/bib/bbz063. Brief Bioinform. 2020. PMID: 31243426 Review.
-
A Single-Cell Sequencing Guide for Immunologists.Front Immunol. 2018 Oct 23;9:2425. doi: 10.3389/fimmu.2018.02425. eCollection 2018. Front Immunol. 2018. PMID: 30405621 Free PMC article. Review.
Cited by
-
LMNA-Related Dilated Cardiomyopathy: Single-Cell Transcriptomics during Patient-Derived iPSC Differentiation Support Cell Type and Lineage-Specific Dysregulation of Gene Expression and Development for Cardiomyocytes and Epicardium-Derived Cells with Lamin A/C Haploinsufficiency.Cells. 2024 Sep 3;13(17):1479. doi: 10.3390/cells13171479. Cells. 2024. PMID: 39273049 Free PMC article.
-
Microglia integration into human midbrain organoids leads to increased neuronal maturation and functionality.Glia. 2022 Jul;70(7):1267-1288. doi: 10.1002/glia.24167. Epub 2022 Mar 9. Glia. 2022. PMID: 35262217 Free PMC article.
-
Single-cell RNA sequencing and binary hierarchical clustering define lung interstitial macrophage heterogeneity in response to hypoxia.Am J Physiol Lung Cell Mol Physiol. 2022 Jul 1;323(1):L58-L68. doi: 10.1152/ajplung.00104.2022. Epub 2022 May 24. Am J Physiol Lung Cell Mol Physiol. 2022. PMID: 35608266 Free PMC article.
-
Characterization of dUTPase expression in mouse postnatal development and adult neurogenesis.Sci Rep. 2024 Jun 7;14(1):13139. doi: 10.1038/s41598-024-63405-0. Sci Rep. 2024. PMID: 38849394 Free PMC article.
-
Single-cell transcriptomics for the assessment of cardiac disease.Nat Rev Cardiol. 2023 May;20(5):289-308. doi: 10.1038/s41569-022-00805-7. Epub 2022 Dec 20. Nat Rev Cardiol. 2023. PMID: 36539452 Review.
