

Genome evolution of SARS-CoV-2 and its virological characteristics
- PMID: 32834891
- PMCID: PMC7415347
- DOI: 10.1186/s41232-020-00126-7
Genome evolution of SARS-CoV-2 and its virological characteristics
Erratum in
-
Correction to: Genome evolution of SARS-CoV-2 and its virological characteristics.Inflamm Regen. 2020 Dec 21;40(1):41. doi: 10.1186/s41232-020-00151-6. Inflamm Regen. 2020. PMID: 33349265 Free PMC article. No abstract available.
Abstract
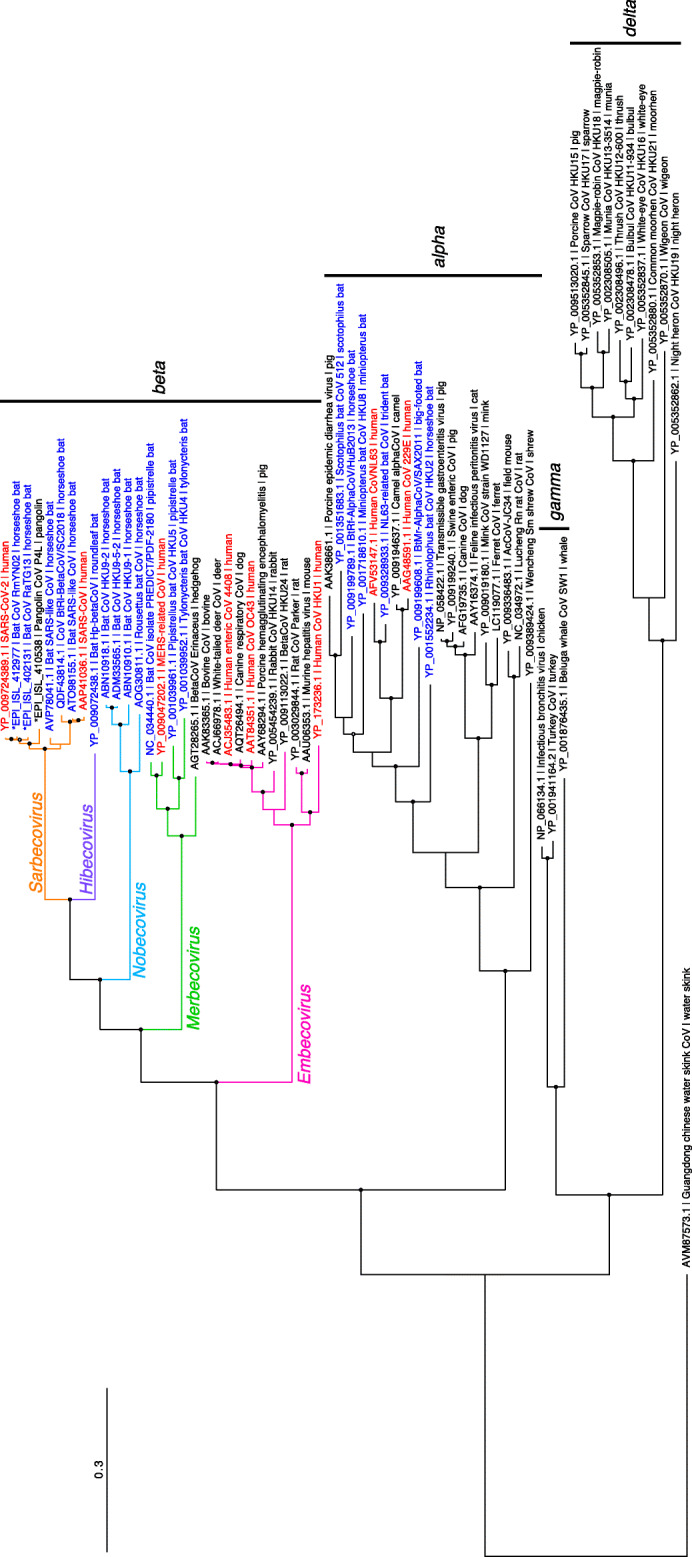
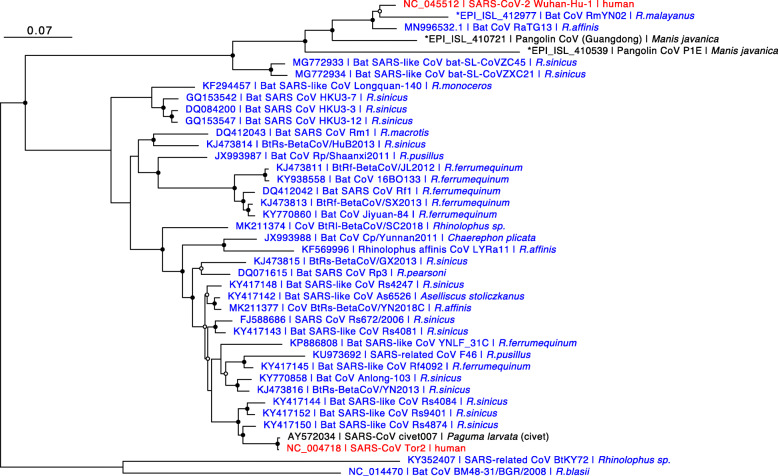
Coronavirus disease of 2019 (COVID-19), which originated in China in 2019, shows mild cold and pneumonia symptoms that can occasionally worsen and result in deaths. SARS-CoV-2 was reported to be the causative agent of the disease and was identified as being similar to SARS-CoV, a causative agent of SARS in 2003. In this review, we described the phylogeny of SARS-CoV-2, covering various related studies, in particular, focusing on viruses obtained from horseshoe bats and pangolins that belong to Sarbecovirus, a subgenus of Betacoronavirus. We also describe the virological characteristics of SARS-CoV-2 and compare them with other coronaviruses. More than 30,000 genome sequences of SARS-CoV-2 are available in the GISAID database as of May 28, 2020. Using the genome sequence data of closely related viruses, the genomic characteristics and evolution of SARS-CoV-2 were extensively studied. However, given the global prevalence of COVID-19 and the large number of associated deaths, further computational and experimental virological analyses are required to fully characterize SARS-CoV-2.
Keywords: COVID-19; Comparative genomics; Coronavirus; SARS-CoV-2; Viral evolution.
© The Author(s) 2020.
Conflict of interest statement
Competing interestsThe authors declare that they have no competing interests.
Figures

Similar articles
-
[Etiology of epidemic outbreaks COVID-19 on Wuhan, Hubei province, Chinese People Republic associated with 2019-nCoV (Nidovirales, Coronaviridae, Coronavirinae, Betacoronavirus, Subgenus Sarbecovirus): lessons of SARS-CoV outbreak.].Vopr Virusol. 2020;65(1):6-15. doi: 10.36233/0507-4088-2020-65-1-6-15. Vopr Virusol. 2020. PMID: 32496715 Review. Russian.
-
Coding potential and sequence conservation of SARS-CoV-2 and related animal viruses.Infect Genet Evol. 2020 Sep;83:104353. doi: 10.1016/j.meegid.2020.104353. Epub 2020 May 5. Infect Genet Evol. 2020. PMID: 32387562 Free PMC article.
-
Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins.Nature. 2020 Jul;583(7815):286-289. doi: 10.1038/s41586-020-2313-x. Epub 2020 May 7. Nature. 2020. PMID: 32380510
-
[Source of the COVID-19 pandemic: ecology and genetics of coronaviruses (Betacoronavirus: Coronaviridae) SARS-CoV, SARS-CoV-2 (subgenus Sarbecovirus), and MERS-CoV (subgenus Merbecovirus).].Vopr Virusol. 2020;65(2):62-70. doi: 10.36233/0507-4088-2020-65-2-62-70. Vopr Virusol. 2020. PMID: 32515561 Review. Russian.
-
Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding.Lancet. 2020 Feb 22;395(10224):565-574. doi: 10.1016/S0140-6736(20)30251-8. Epub 2020 Jan 30. Lancet. 2020. PMID: 32007145 Free PMC article.
Cited by
-
Virtual screening based on the structure of more than 105 compounds against four key proteins of SARS-CoV-2: MPro, SRBD, RdRp, and PLpro.Inform Med Unlocked. 2022;35:101134. doi: 10.1016/j.imu.2022.101134. Epub 2022 Nov 12. Inform Med Unlocked. 2022. PMID: 36406927 Free PMC article.
-
Sarbecovirus ORF6 proteins hamper induction of interferon signaling.Cell Rep. 2021 Mar 30;34(13):108916. doi: 10.1016/j.celrep.2021.108916. Epub 2021 Mar 12. Cell Rep. 2021. PMID: 33765414 Free PMC article.
-
Impact of SARS-CoV-2 on pregnancy outcomes (Review).Med Int (Lond). 2021 Oct 26;1(5):19. doi: 10.3892/mi.2021.19. eCollection 2021 Nov-Dec. Med Int (Lond). 2021. PMID: 36698529 Free PMC article. Review.
-
SARS-CoV-2 Within-Host and in vitro Genomic Variability and Sub-Genomic RNA Levels Indicate Differences in Viral Expression Between Clinical Cohorts and in vitro Culture.Front Microbiol. 2022 May 19;13:824217. doi: 10.3389/fmicb.2022.824217. eCollection 2022. Front Microbiol. 2022. PMID: 35663867 Free PMC article.
-
Origin, phylogeny, variability and epitope conservation of SARS-CoV-2 worldwide.Virus Res. 2021 Oct 15;304:198526. doi: 10.1016/j.virusres.2021.198526. Epub 2021 Jul 30. Virus Res. 2021. PMID: 34339772 Free PMC article.
References
-
- WHO (World Health Organization): Novel Coronavirus (2019-nCoV) situation report – 22; https://www.who.int/docs/default-source/coronaviruse/situation-reports/2.... (Accessed 25 May 2020).
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
