

Compensatory Estrogen Signal Is Capable of DNA Repair in Antiestrogen-Responsive Cancer Cells via Activating Mutations
- PMID: 32774370
- PMCID: PMC7407016
- DOI: 10.1155/2020/5418365
Compensatory Estrogen Signal Is Capable of DNA Repair in Antiestrogen-Responsive Cancer Cells via Activating Mutations
Abstract
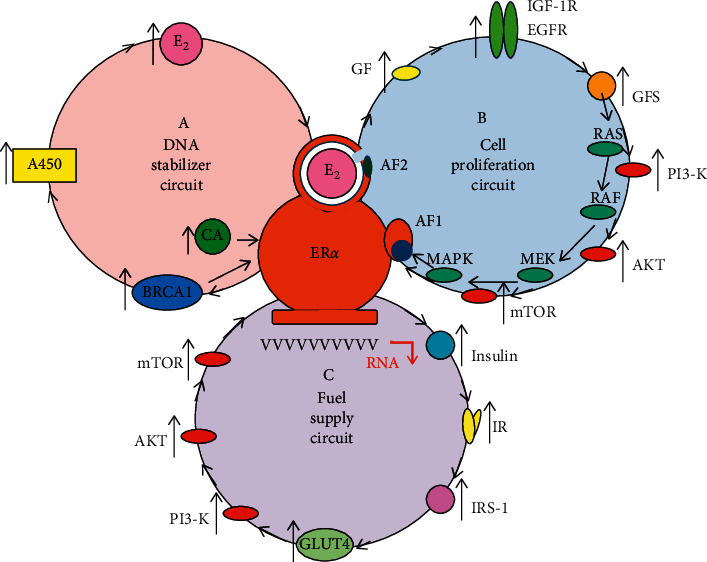
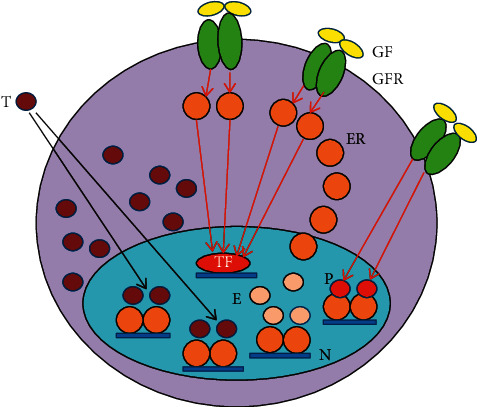
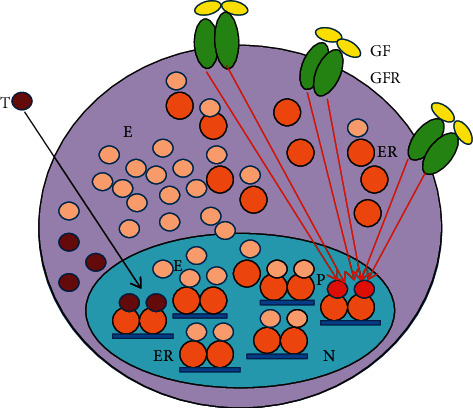
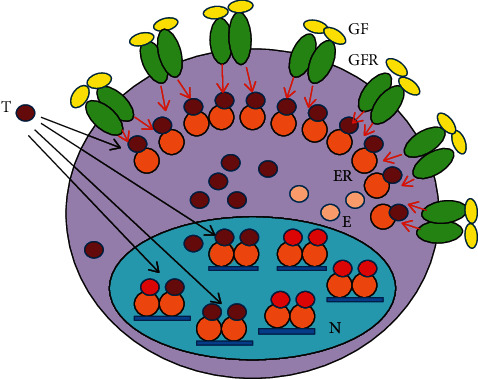
Cancer cells are embarrassed human cells exhibiting the remnants of same mechanisms for DNA stabilization like patients have in their healthy cells. Antiestrogens target the liganded activation of ERs, which is the principal means of genomic regulation in both patients and their tumors. The artificial blockade of liganded ER activation is an emergency situation promoting strong compensatory actions even in cancer cells. When tumor cells are capable of an appropriate upregulation of ER signaling resulting in DNA repair, a tumor response may be detected. In contrast, when ER signaling is completely inhibited, tumor cells show unrestrained proliferation, and tumor growth may be observed. The laboratory investigations of genomic mechanisms in antiestrogen-responsive and antiestrogen-unresponsive tumor cells have considerably enhanced our knowledge regarding the principal regulatory capacity of estrogen signaling. In antiestrogen-responsive tumor cells, a compensatory increased expression and liganded activation of estrogen receptors (ERs) result in an apoptotic death. Conversely, in antiestrogen resistant tumors exhibiting a complete blockade of liganded ER activation, a compensatory effort for unliganded ER activation is characteristic, conferred by the increased expression and activity of growth factor receptors. However, even extreme unliganded ER activation is incapable of DNA restoration when the liganded ER activation is completely blocked. Researchers mistakenly suspect even today that in tumors growing under antiestrogen treatment, the increased unliganded activation of estrogen receptor via activating mutations is an aggressive survival technique, whilst it is a compensatory effort against the blockade of liganded ER activation. The capacity of liganded ERs for genome modification in emergency states provides possibilities for estrogen/ER use in medical practice including cancer cure.
Copyright © 2020 Zsuzsanna Suba.
Conflict of interest statement
The author declares that there are no conflicts of interest.
Figures
Similar articles
-
Rosetta Stone for Cancer Cure: Comparison of the Anticancer Capacity of Endogenous Estrogens, Synthetic Estrogens and Antiestrogens.Oncol Rev. 2023 Apr 19;17:10708. doi: 10.3389/or.2023.10708. eCollection 2023. Oncol Rev. 2023. PMID: 37152665 Free PMC article. Review.
-
DNA Damage Responses in Tumors Are Not Proliferative Stimuli, but Rather They Are DNA Repair Actions Requiring Supportive Medical Care.Cancers (Basel). 2024 Apr 19;16(8):1573. doi: 10.3390/cancers16081573. Cancers (Basel). 2024. PMID: 38672654 Free PMC article. Review.
-
Activating Mutations of ESR1, BRCA1 and CYP19 Aromatase Genes Confer Tumor Response in Breast Cancers Treated with Antiestrogens.Recent Pat Anticancer Drug Discov. 2017;12(2):136-147. doi: 10.2174/1574892812666170227110842. Recent Pat Anticancer Drug Discov. 2017. PMID: 28245776 Review.
-
Amplified Crosstalk Between Estrogen Binding and GFR Signaling Mediated Pathways of ER Activation Drives Responses in Tumors Treated with Endocrine Disruptors.Recent Pat Anticancer Drug Discov. 2018;13(4):428-444. doi: 10.2174/1574892813666180720123732. Recent Pat Anticancer Drug Discov. 2018. PMID: 30027855 Review.
-
The pitfall of the transient, inconsistent anticancer capacity of antiestrogens and the mechanism of apparent antiestrogen resistance.Drug Des Devel Ther. 2015 Aug 6;9:4341-53. doi: 10.2147/DDDT.S89536. eCollection 2015. Drug Des Devel Ther. 2015. PMID: 26273195 Free PMC article.
Cited by
-
Estrogen Regulated Genes Compel Apoptosis in Breast Cancer Cells, Whilst Stimulate Antitumor Activity in Peritumoral Immune Cells in a Janus-Faced Manner.Curr Oncol. 2024 Aug 24;31(9):4885-4907. doi: 10.3390/curroncol31090362. Curr Oncol. 2024. PMID: 39329990 Free PMC article. Review.
-
Rosetta Stone for Cancer Cure: Comparison of the Anticancer Capacity of Endogenous Estrogens, Synthetic Estrogens and Antiestrogens.Oncol Rev. 2023 Apr 19;17:10708. doi: 10.3389/or.2023.10708. eCollection 2023. Oncol Rev. 2023. PMID: 37152665 Free PMC article. Review.
-
DNA Damage Responses in Tumors Are Not Proliferative Stimuli, but Rather They Are DNA Repair Actions Requiring Supportive Medical Care.Cancers (Basel). 2024 Apr 19;16(8):1573. doi: 10.3390/cancers16081573. Cancers (Basel). 2024. PMID: 38672654 Free PMC article. Review.
-
A Cohort Study Investigating Zearalenone Concentrations and Selected Steroid Levels in Patients with Sigmoid Colorectal Cancer or Colorectal Cancer.Toxins (Basel). 2023 Dec 27;16(1):15. doi: 10.3390/toxins16010015. Toxins (Basel). 2023. PMID: 38251232 Free PMC article.
-
Estrogen and Progesterone Receptors Are Dysregulated at the BPH/5 Mouse Preeclamptic-Like Maternal-Fetal Interface.Biology (Basel). 2024 Mar 16;13(3):192. doi: 10.3390/biology13030192. Biology (Basel). 2024. PMID: 38534461 Free PMC article.
References
-
- Suba Z. Topics in Anti-Cancer Research. Vol. 8. New York, NY, USA: Bentham Science Publishers; 2019. Synthetic estrogens deregulate estrogen receptors inducing thromboembolic complications and cancer; pp. 44–73.
-
- Harvey J. M., Clark G. M., Osborne C. K., Allred D. C. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. Journal of Clinical Oncology. 1999;17(5):p. 1474. doi: 10.1200/jco.1999.17.5.1474. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
