

Validation of Variant Assembly Using HAPHPIPE with Next-Generation Sequence Data from Viruses
- PMID: 32674515
- PMCID: PMC7412389
- DOI: 10.3390/v12070758
Validation of Variant Assembly Using HAPHPIPE with Next-Generation Sequence Data from Viruses
Abstract
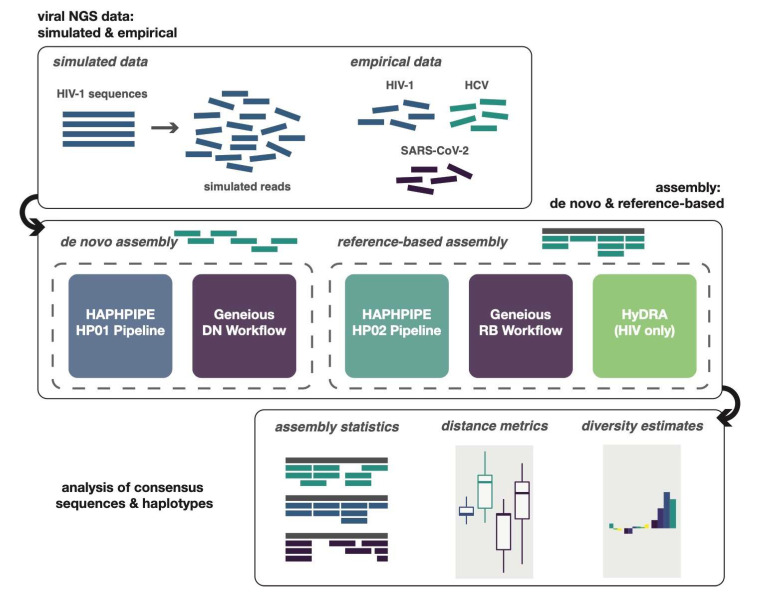
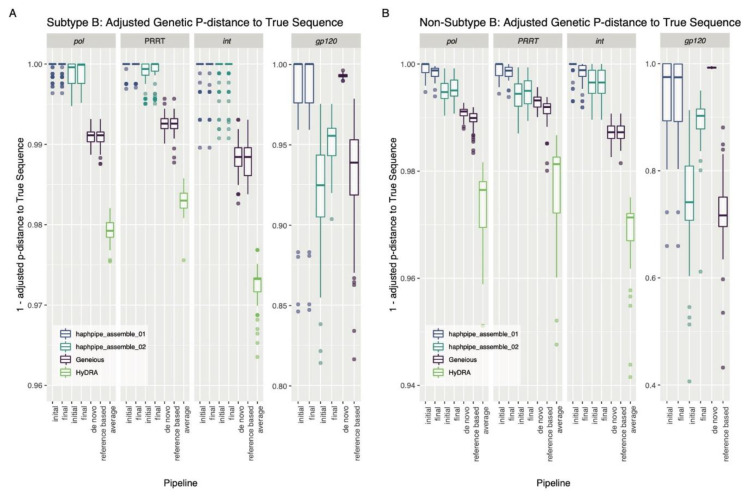
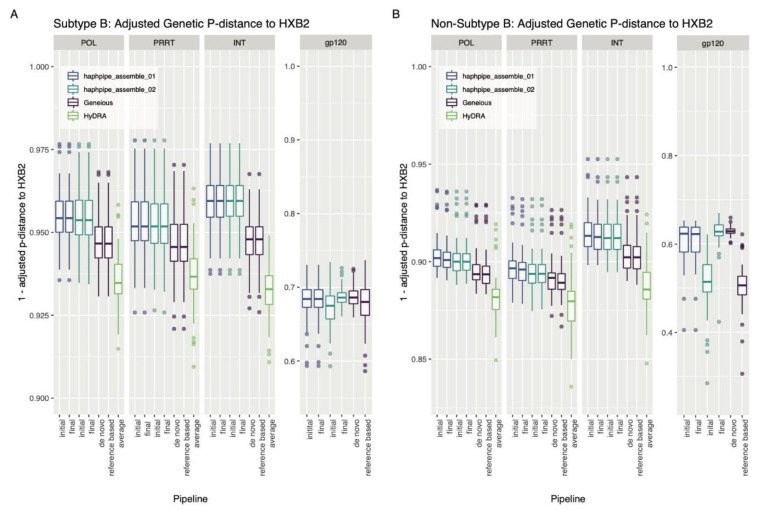
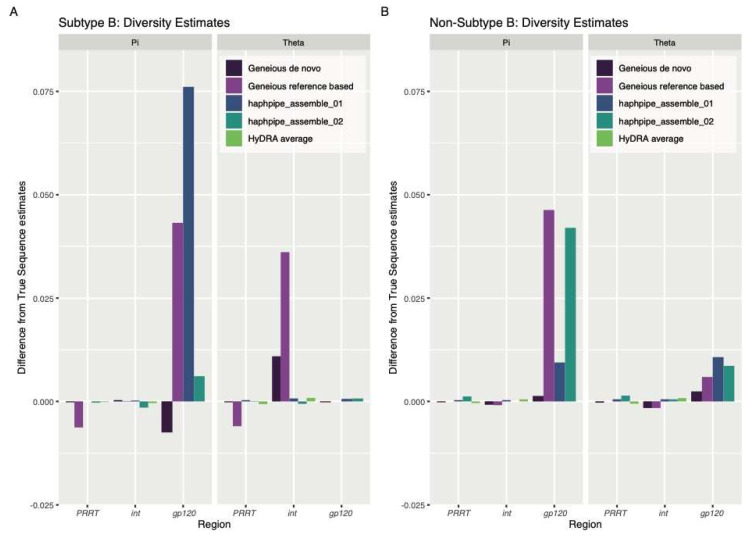
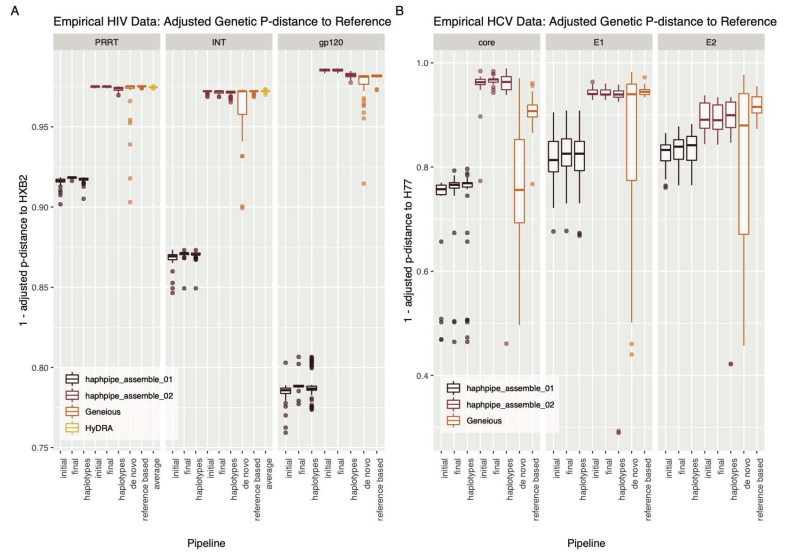
Next-generation sequencing (NGS) offers a powerful opportunity to identify low-abundance, intra-host viral sequence variants, yet the focus of many bioinformatic tools on consensus sequence construction has precluded a thorough analysis of intra-host diversity. To take full advantage of the resolution of NGS data, we developed HAplotype PHylodynamics PIPEline (HAPHPIPE), an open-source tool for the de novo and reference-based assembly of viral NGS data, with both consensus sequence assembly and a focus on the quantification of intra-host variation through haplotype reconstruction. We validate and compare the consensus sequence assembly methods of HAPHPIPE to those of two alternative software packages, HyDRA and Geneious, using simulated HIV and empirical HIV, HCV, and SARS-CoV-2 datasets. Our validation methods included read mapping, genetic distance, and genetic diversity metrics. In simulated NGS data, HAPHPIPE generated pol consensus sequences significantly closer to the true consensus sequence than those produced by HyDRA and Geneious and performed comparably to Geneious for HIV gp120 sequences. Furthermore, using empirical data from multiple viruses, we demonstrate that HAPHPIPE can analyze larger sequence datasets due to its greater computational speed. Therefore, we contend that HAPHPIPE provides a more user-friendly platform for users with and without bioinformatics experience to implement current best practices for viral NGS assembly than other currently available options.
Keywords: HCV; HIV; SARS-CoV-2; bioinformatics; consensus; haplotypes; simulation; validation; viruses.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
HAPHPIPE: Haplotype Reconstruction and Phylodynamics for Deep Sequencing of Intrahost Viral Populations.Mol Biol Evol. 2021 Apr 13;38(4):1677-1690. doi: 10.1093/molbev/msaa315. Mol Biol Evol. 2021. PMID: 33367849 Free PMC article.
-
Evaluation of haplotype callers for next-generation sequencing of viruses.Infect Genet Evol. 2020 Aug;82:104277. doi: 10.1016/j.meegid.2020.104277. Epub 2020 Mar 6. Infect Genet Evol. 2020. PMID: 32151775 Free PMC article.
-
An online coronavirus analysis platform from the National Genomics Data Center.Zool Res. 2020 Nov 18;41(6):705-708. doi: 10.24272/j.issn.2095-8137.2020.065. Zool Res. 2020. PMID: 33045776 Free PMC article.
-
Bioinformatics tools for analysing viral genomic data.Rev Sci Tech. 2016 Apr;35(1):271-85. doi: 10.20506/rst.35.1.2432. Rev Sci Tech. 2016. PMID: 27217183 Review.
-
Applications of next-generation sequencing technologies to diagnostic virology.Int J Mol Sci. 2011;12(11):7861-84. doi: 10.3390/ijms12117861. Epub 2011 Nov 14. Int J Mol Sci. 2011. PMID: 22174638 Free PMC article. Review.
Cited by
-
Altered RSV Epidemiology and Genetic Diversity Following the COVID-19 Pandemic.Res Sq [Preprint]. 2023 Dec 15:rs.3.rs-3712859. doi: 10.21203/rs.3.rs-3712859/v1. Res Sq. 2023. Update in: Nat Commun. 2024 Apr 20;15(1):3374. doi: 10.1038/s41467-024-47757-9. PMID: 38168164 Free PMC article. Updated. Preprint.
-
HAPHPIPE: Haplotype Reconstruction and Phylodynamics for Deep Sequencing of Intrahost Viral Populations.Mol Biol Evol. 2021 Apr 13;38(4):1677-1690. doi: 10.1093/molbev/msaa315. Mol Biol Evol. 2021. PMID: 33367849 Free PMC article.
-
ViralWasm: a client-side user-friendly web application suite for viral genomics.Bioinformatics. 2024 Jan 2;40(1):btae018. doi: 10.1093/bioinformatics/btae018. Bioinformatics. 2024. PMID: 38200583 Free PMC article.
-
Deviations in RSV epidemiological patterns and population structures in the United States following the COVID-19 pandemic.Nat Commun. 2024 Apr 20;15(1):3374. doi: 10.1038/s41467-024-47757-9. Nat Commun. 2024. PMID: 38643200 Free PMC article.
References
-
- Bonnaud E.M., Troupin C., Dacheux L., Holmes E.C., Monchatre-Leroy E., Tanguy M., Bouchier C., Cliquet F., Barrat J., Bourhy H. Comparison of intra- and inter-host genetic diversity in rabies virus during experimental cross-species transmission. PLOS Pathog. 2019;15:e1007799. doi: 10.1371/journal.ppat.1007799. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
