

Innate Immune Sensing of Influenza A Virus
- PMID: 32674269
- PMCID: PMC7411791
- DOI: 10.3390/v12070755
Innate Immune Sensing of Influenza A Virus
Abstract
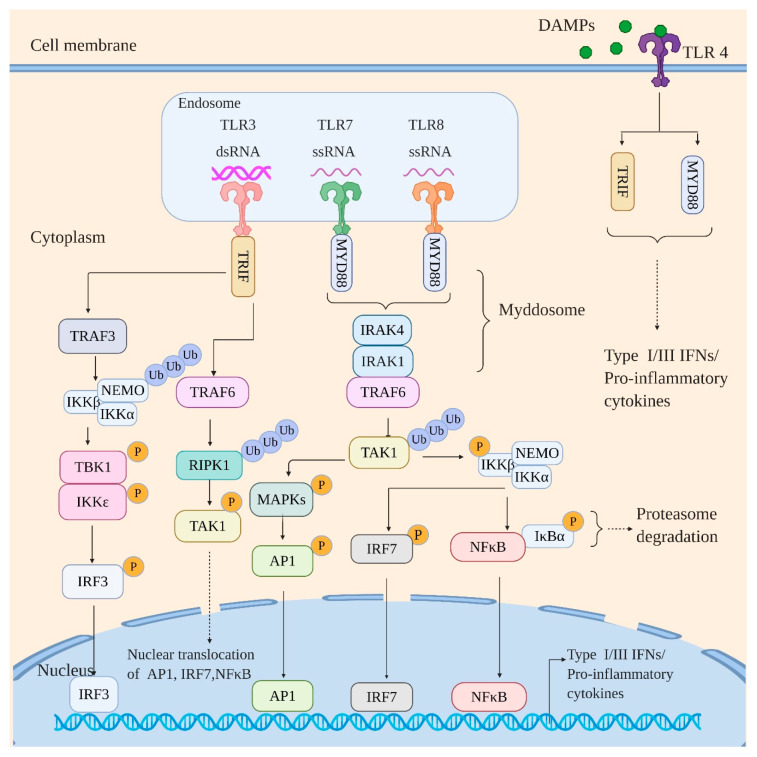
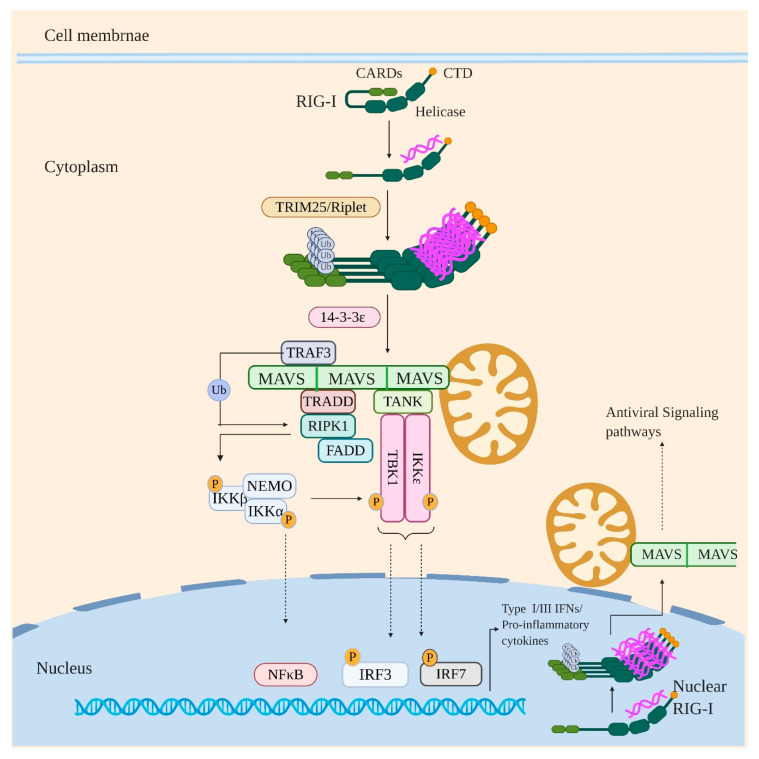
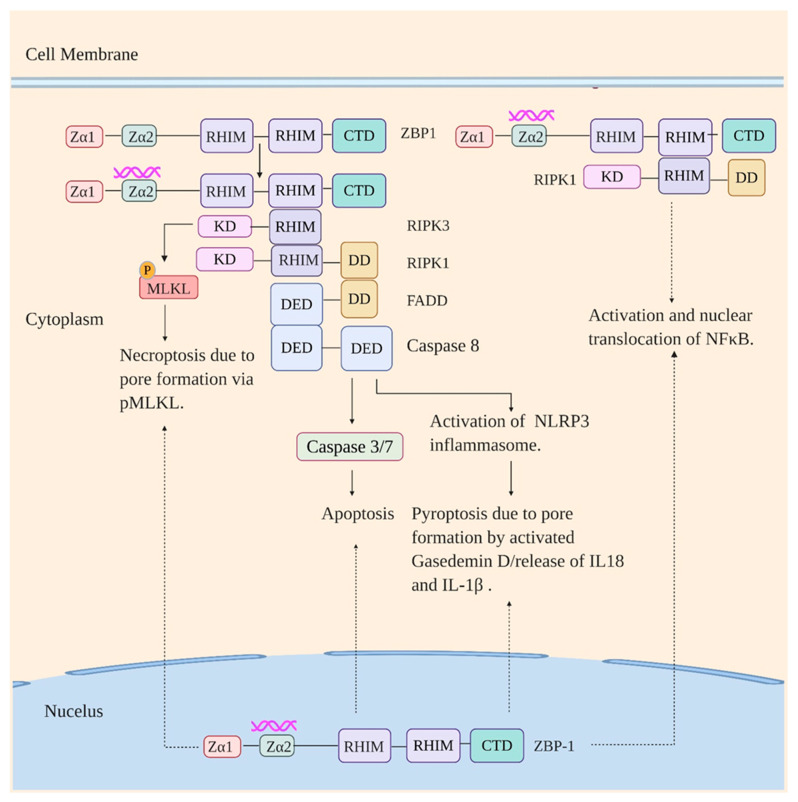
Influenza virus infection triggers host innate immune response by stimulating various pattern recognition receptors (PRRs). Activation of these PRRs leads to the activation of a plethora of signaling pathways, resulting in the production of interferon (IFN) and proinflammatory cytokines, followed by the expression of interferon-stimulated genes (ISGs), the recruitment of innate immune cells, or the activation of programmed cell death. All these antiviral approaches collectively restrict viral replication inside the host. However, influenza virus also engages in multiple mechanisms to subvert the innate immune responses. In this review, we discuss the role of PRRs such as Toll-like receptors (TLRs), Retinoic acid-inducible gene I (RIG-I), NOD-, LRR-, pyrin domain-containing protein 3 (NLRP3), and Z-DNA binding protein 1 (ZBP1) in sensing and restricting influenza viral infection. Further, we also discuss the mechanisms influenza virus utilizes, especially the role of viral non-structure proteins NS1, PB1-F2, and PA-X, to evade the host innate immune responses.
Keywords: influenza virus; innate immune response; pattern recognition receptors.
Conflict of interest statement
The authors declare no conflict of interest. This article is published with the permission of the VIDO-InterVac Director and was assigned a manuscript no. 901.
Figures

Similar articles
-
[Mechanisms underlying interferon-mediated host innate immunity during influenza A virus infection].Sheng Wu Gong Cheng Xue Bao. 2015 Dec;31(12):1671-81. Sheng Wu Gong Cheng Xue Bao. 2015. PMID: 27093830 Review. Chinese.
-
The role of the NLRP3 inflammasome in regulation of antiviral responses to influenza A virus infection.Antiviral Res. 2017 Dec;148:32-42. doi: 10.1016/j.antiviral.2017.10.020. Epub 2017 Oct 31. Antiviral Res. 2017. PMID: 29097227 Review.
-
Virus sensing receptors in cellular infectivity of influenza A virus.J Infect Dev Ctries. 2021 Jan 31;15(1):1-8. doi: 10.3855/jidc.13258. J Infect Dev Ctries. 2021. PMID: 33571140 Review.
-
Recognition of Viral RNA by Pattern Recognition Receptors in the Induction of Innate Immunity and Excessive Inflammation During Respiratory Viral Infections.Viral Immunol. 2017 Jul/Aug;30(6):408-420. doi: 10.1089/vim.2016.0178. Epub 2017 Jun 13. Viral Immunol. 2017. PMID: 28609250 Review.
-
Modulation of Innate Immune Responses by the Influenza A NS1 and PA-X Proteins.Viruses. 2018 Dec 12;10(12):708. doi: 10.3390/v10120708. Viruses. 2018. PMID: 30545063 Free PMC article. Review.
Cited by
-
Regulation of Antiviral Immune Response by N 6-Methyladenosine of mRNA.Front Microbiol. 2021 Dec 16;12:789605. doi: 10.3389/fmicb.2021.789605. eCollection 2021. Front Microbiol. 2021. PMID: 34975810 Free PMC article. Review.
-
Multi-spectral Fluorescent Reporter Influenza A Viruses Allow for in vivo Studies of Innate Immune Function in Zebrafish.bioRxiv [Preprint]. 2023 Nov 2:2023.10.31.564888. doi: 10.1101/2023.10.31.564888. bioRxiv. 2023. Update in: Viruses. 2024 Jan 20;16(1):155. doi: 10.3390/v16010155 PMID: 37961402 Free PMC article. Updated. Preprint.
-
Co-expression network analysis identifies potential candidate hub genes in severe influenza patients needing invasive mechanical ventilation.BMC Genomics. 2022 Oct 15;23(1):703. doi: 10.1186/s12864-022-08915-9. BMC Genomics. 2022. PMID: 36243706 Free PMC article.
-
Strategies of Influenza A Virus to Ensure the Translation of Viral mRNAs.Pathogens. 2022 Dec 12;11(12):1521. doi: 10.3390/pathogens11121521. Pathogens. 2022. PMID: 36558855 Free PMC article. Review.
-
The immune system of chicken and its response to H9N2 avian influenza virus.Vet Q. 2023 Dec;43(1):1-14. doi: 10.1080/01652176.2023.2228360. Vet Q. 2023. PMID: 37357919 Free PMC article. Review.
References
-
- De Vries E., Tscherne D.M., Wienholts M.J., Cobos-Jiménez V., Scholte F., García-Sastre A., Rottier P.J.M., de Haan C.A.M. Dissection of the Influenza A Virus Endocytic Routes Reveals Macropinocytosis as an Alternative Entry Pathway. PLoS Pathog. 2011;7:e1001329. doi: 10.1371/journal.ppat.1001329. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
