

Integrated Transcriptome and Network Analysis Reveals Spatiotemporal Dynamics of Calvarial Suturogenesis
- PMID: 32640236
- PMCID: PMC7379176
- DOI: 10.1016/j.celrep.2020.107871
Integrated Transcriptome and Network Analysis Reveals Spatiotemporal Dynamics of Calvarial Suturogenesis
Abstract
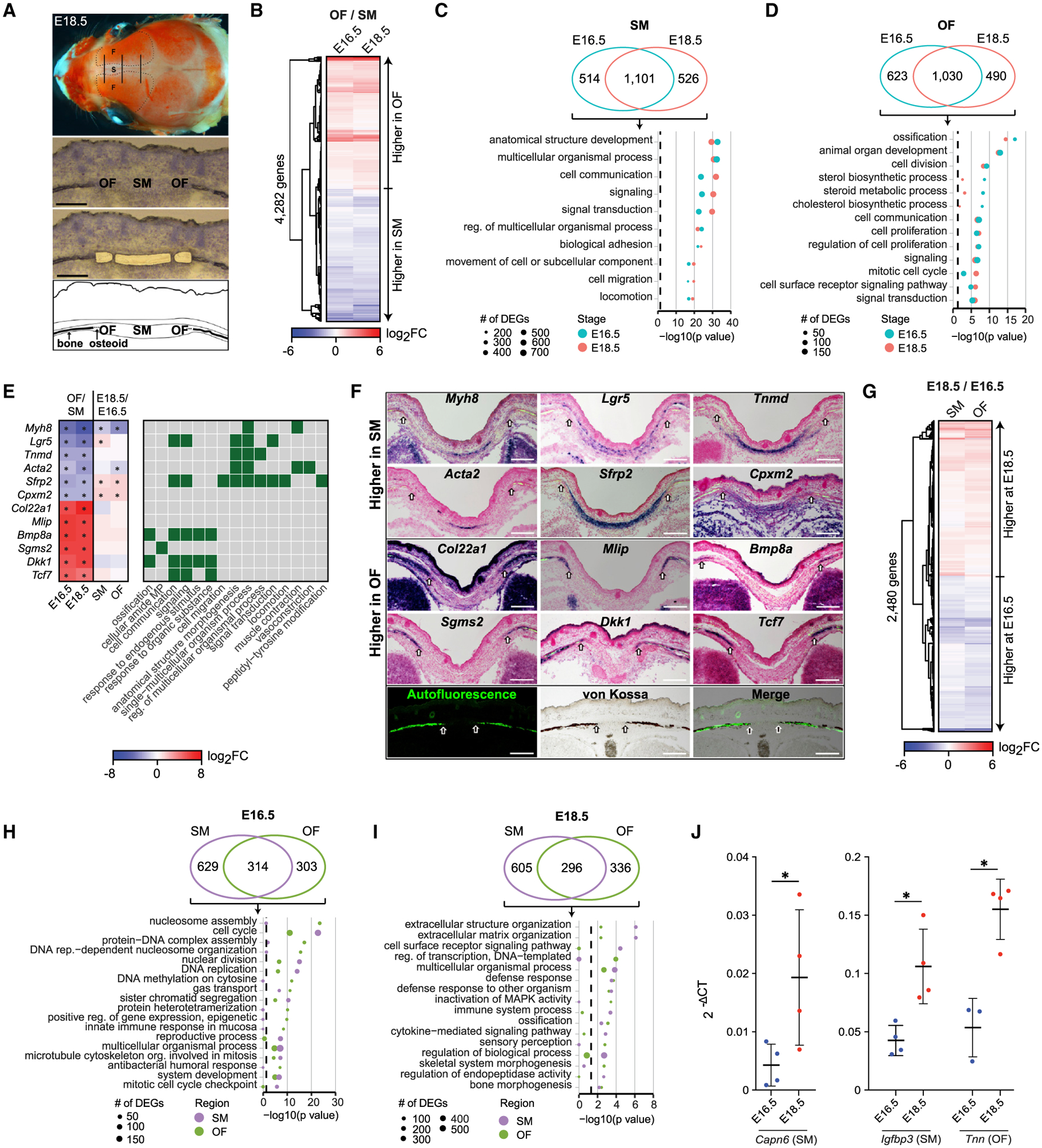
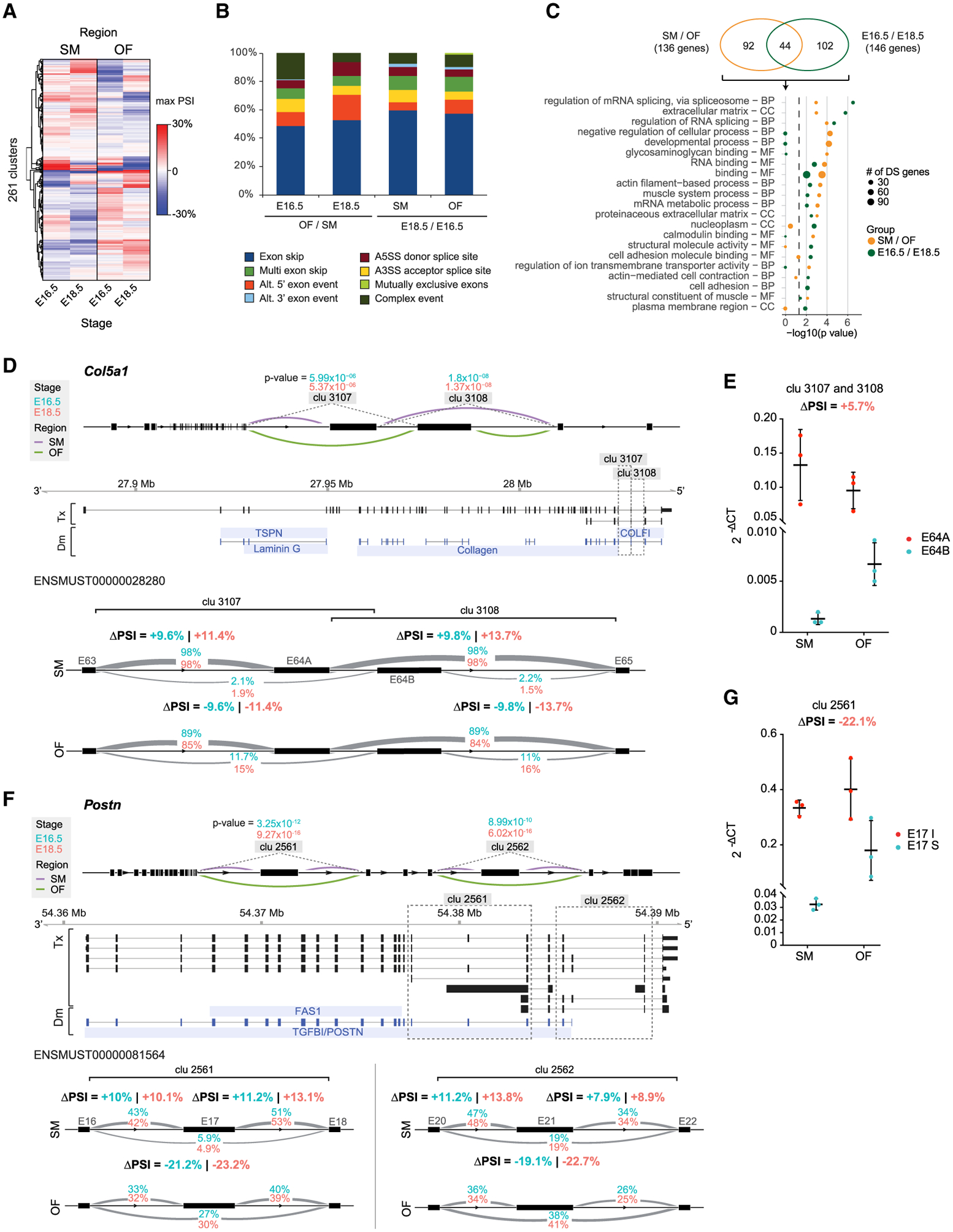
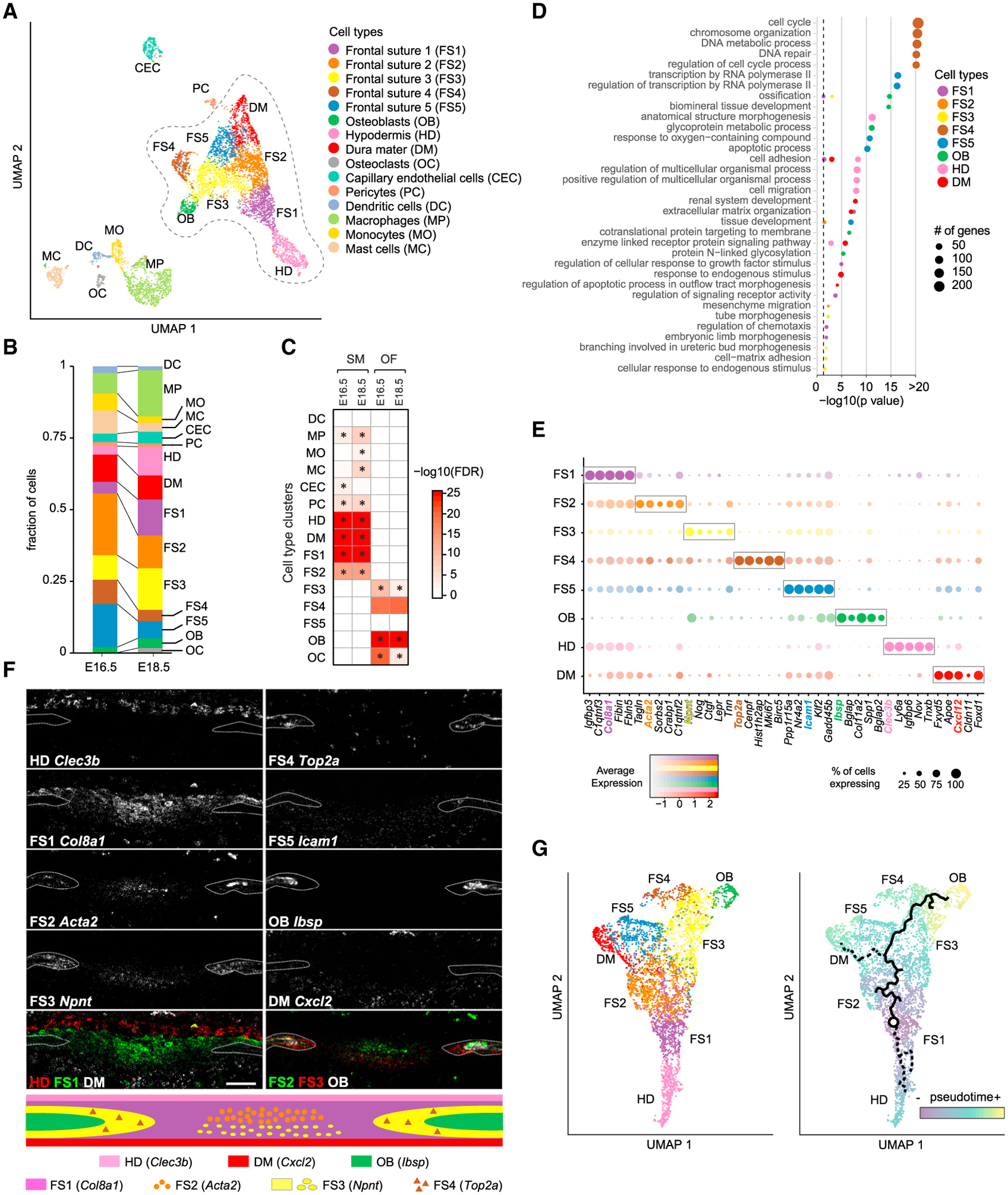
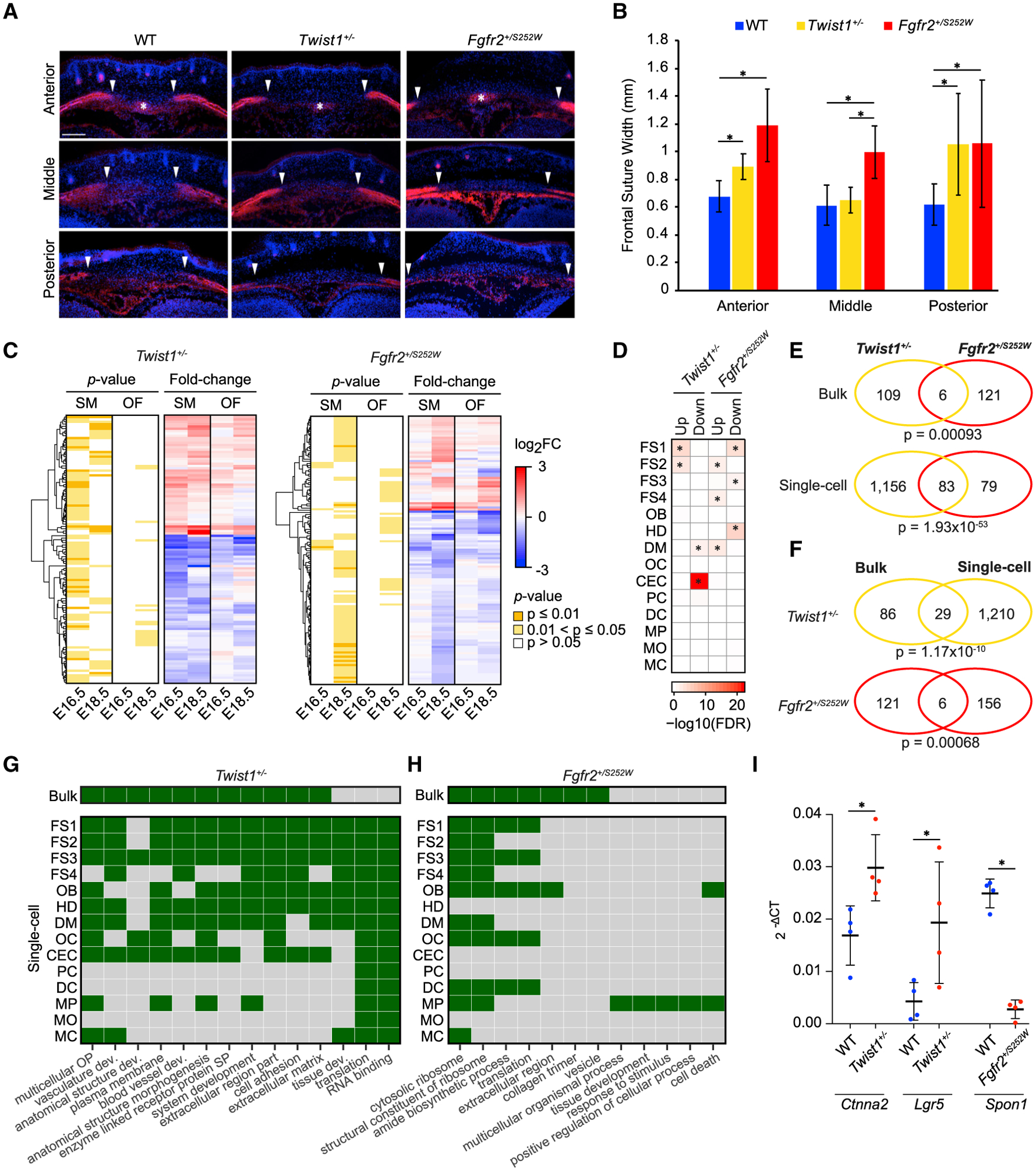
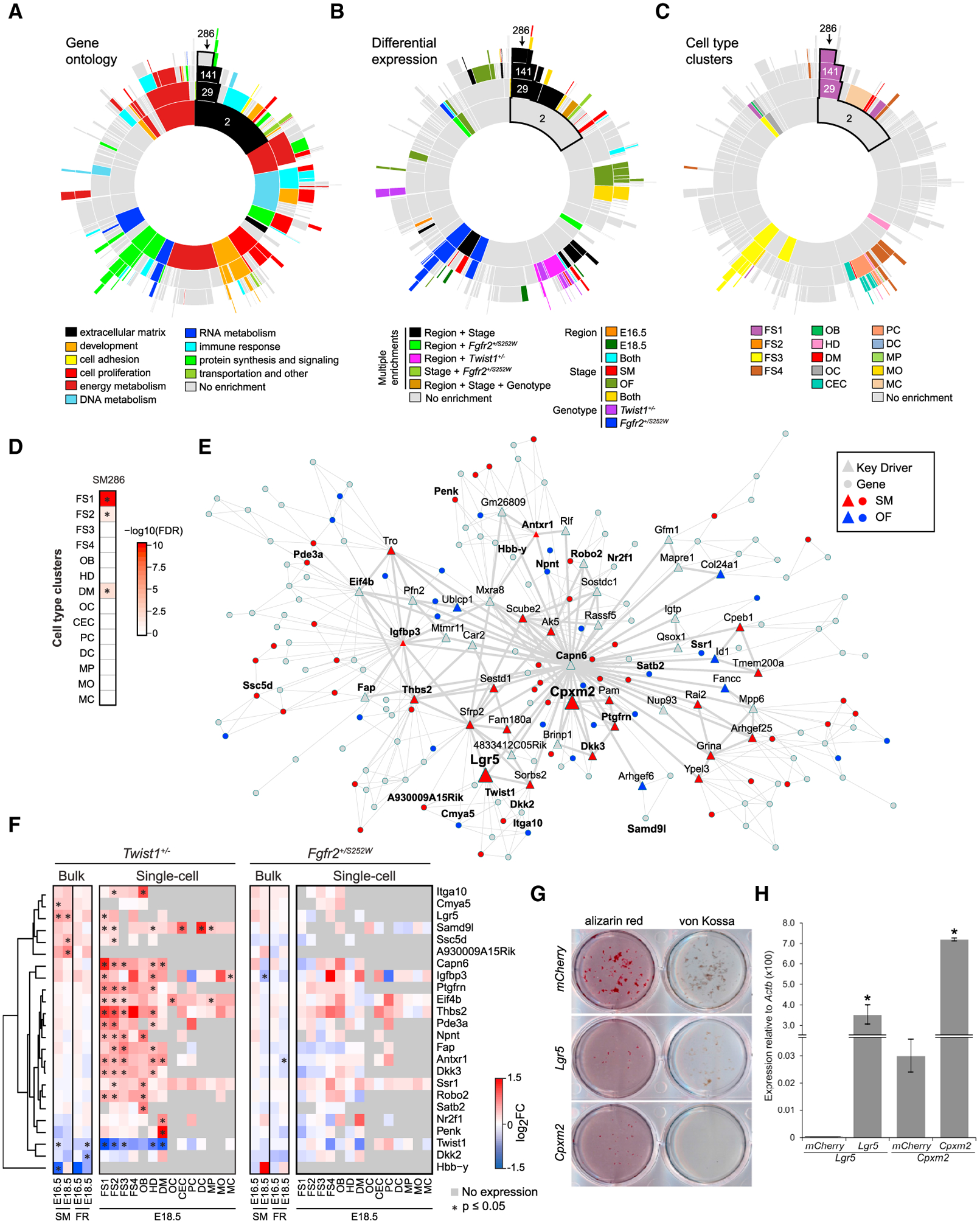
Craniofacial abnormalities often involve sutures, the growth centers of the skull. To characterize the organization and processes governing their development, we profile the murine frontal suture, a model for sutural growth and fusion, at the tissue- and single-cell level on embryonic days (E)16.5 and E18.5. For the wild-type suture, bulk RNA sequencing (RNA-seq) analysis identifies mesenchyme-, osteogenic front-, and stage-enriched genes and biological processes, as well as alternative splicing events modifying the extracellular matrix. Single-cell RNA-seq analysis distinguishes multiple subpopulations, of which five define a mesenchyme-osteoblast differentiation trajectory and show variation along the anteroposterior axis. Similar analyses of in vivo mouse models of impaired frontal suturogenesis in Saethre-Chotzen and Apert syndromes, Twist1+/- and Fgfr2+/S252W, demonstrate distinct transcriptional changes involving angiogenesis and ribogenesis, respectively. Co-expression network analysis reveals gene expression modules from which we validate key driver genes regulating osteoblast differentiation. Our study provides a global approach to gain insights into suturogenesis.
Keywords: Fgfr2; Twist1; bone; craniofacial; craniosynostosis; differential gene expression; frontal suture; mesenchyme; metopic suture; single-cell RNA-seq.
Copyright © 2020 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
Similar articles
-
The developing mouse coronal suture at single-cell resolution.Nat Commun. 2021 Aug 10;12(1):4797. doi: 10.1038/s41467-021-24917-9. Nat Commun. 2021. PMID: 34376651 Free PMC article.
-
Mesodermal expression of Fgfr2S252W is necessary and sufficient to induce craniosynostosis in a mouse model of Apert syndrome.Dev Biol. 2012 Aug 15;368(2):283-93. doi: 10.1016/j.ydbio.2012.05.026. Epub 2012 Jun 1. Dev Biol. 2012. PMID: 22664175 Free PMC article.
-
Single-cell analysis identifies a key role for Hhip in murine coronal suture development.Nat Commun. 2021 Dec 8;12(1):7132. doi: 10.1038/s41467-021-27402-5. Nat Commun. 2021. PMID: 34880220 Free PMC article.
-
The Development of the Calvarial Bones and Sutures and the Pathophysiology of Craniosynostosis.Curr Top Dev Biol. 2015;115:131-56. doi: 10.1016/bs.ctdb.2015.07.004. Epub 2015 Oct 1. Curr Top Dev Biol. 2015. PMID: 26589924 Review.
-
Developmental anatomy of craniofacial sutures.Front Oral Biol. 2008;12:1-21. doi: 10.1159/000115028. Front Oral Biol. 2008. PMID: 18391492 Review.
Cited by
-
Cranial Neural Crest Cells and Their Role in the Pathogenesis of Craniofacial Anomalies and Coronal Craniosynostosis.J Dev Biol. 2020 Sep 9;8(3):18. doi: 10.3390/jdb8030018. J Dev Biol. 2020. PMID: 32916911 Free PMC article. Review.
-
Craniofacial developmental biology in the single-cell era.Development. 2023 Oct 1;150(19):dev202077. doi: 10.1242/dev.202077. Epub 2023 Oct 9. Development. 2023. PMID: 37812056 Free PMC article. Review.
-
Erf Affects Commitment and Differentiation of Osteoprogenitor Cells in Cranial Sutures via the Retinoic Acid Pathway.Mol Cell Biol. 2021 Jul 23;41(8):e0014921. doi: 10.1128/MCB.00149-21. Epub 2021 Jul 23. Mol Cell Biol. 2021. PMID: 33972395 Free PMC article.
-
Cranium growth, patterning and homeostasis.Development. 2022 Nov 15;149(22):dev201017. doi: 10.1242/dev.201017. Epub 2022 Nov 21. Development. 2022. PMID: 36408946 Free PMC article.
-
The molecular complex of ciliary and golgin protein is crucial for skull development.Development. 2021 Jul 1;148(13):dev199559. doi: 10.1242/dev.199559. Epub 2021 Jul 1. Development. 2021. PMID: 34128978 Free PMC article.
References
-
- Aslan H, Ravid-Amir O, Clancy BM, Rezvankhah S, Pittman D, Pelled G, Turgeman G, Zilberman Y, Gazit Z, Hoffmann A, et al. (2006). Advanced molecular profiling in vivo detects novel function of dickkopf-3 in the regulation of bone formation. J. Bone Miner. Res 21, 1935–1945. - PubMed
-
- Bean CJ, Hunt PA, Millie EA, and Hassold TJ (2001). Analysis of a mal-segregating mouse Y chromosome: evidence that the earliest cleavage divisions of the mammalian embryo are non-disjunction-prone. Hum. Mol. Genet 10, 963–972. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
