

NF-κB-Interacting Long Noncoding RNA Regulates HIV-1 Replication and Latency by Repressing NF-κB Signaling
- PMID: 32581100
- PMCID: PMC7431781
- DOI: 10.1128/JVI.01057-20
NF-κB-Interacting Long Noncoding RNA Regulates HIV-1 Replication and Latency by Repressing NF-κB Signaling
Abstract
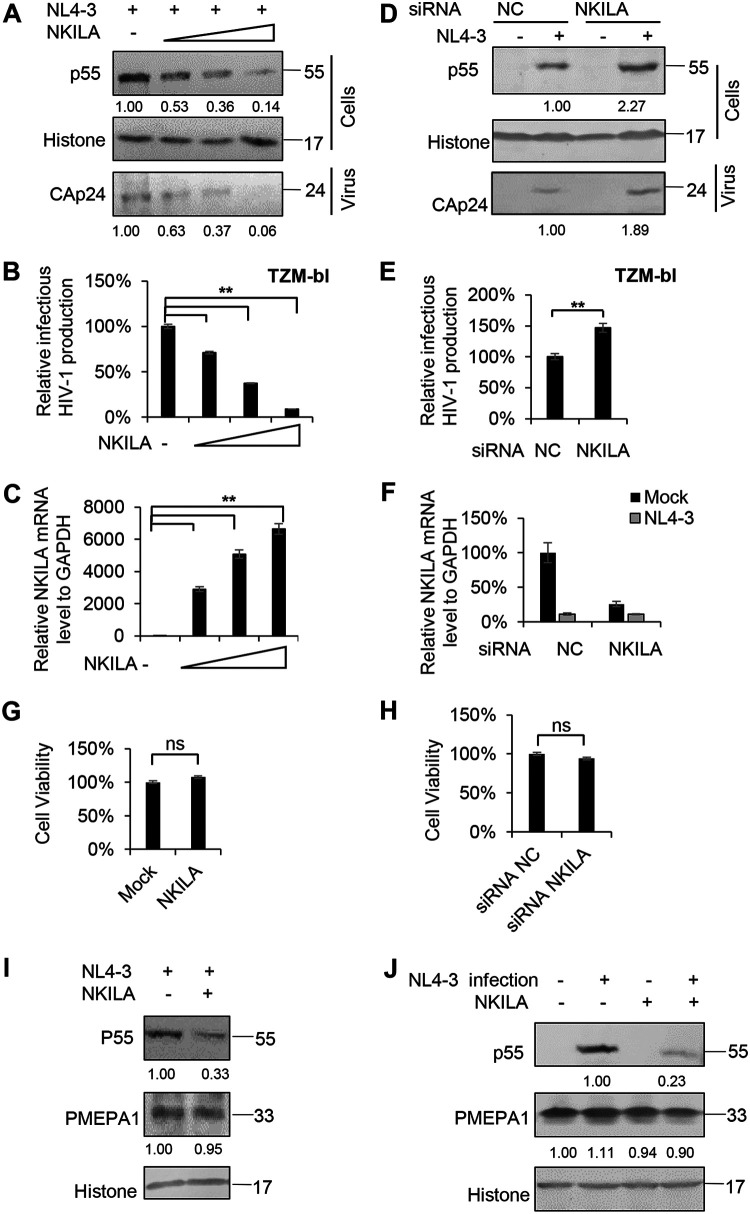
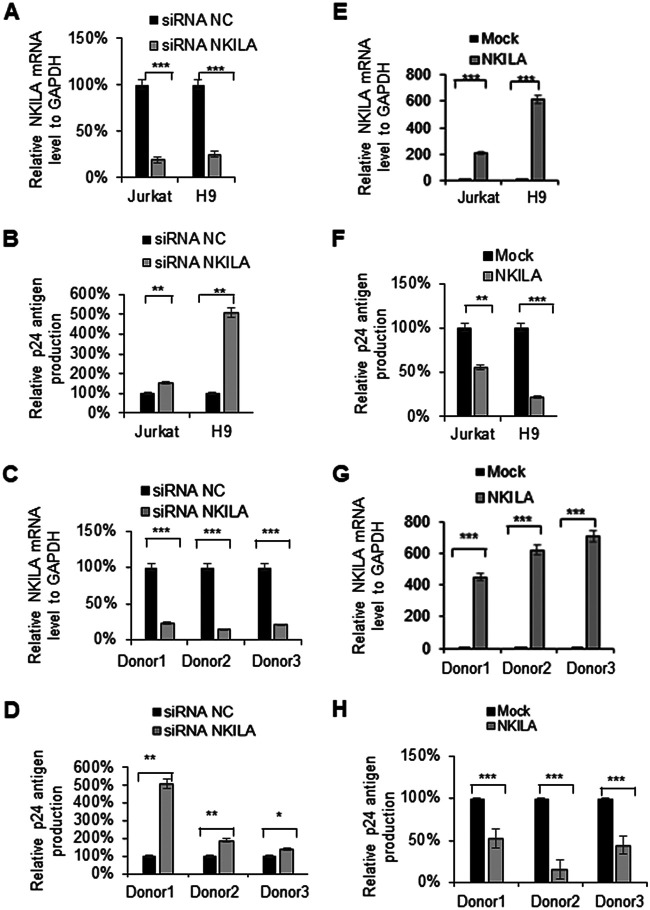
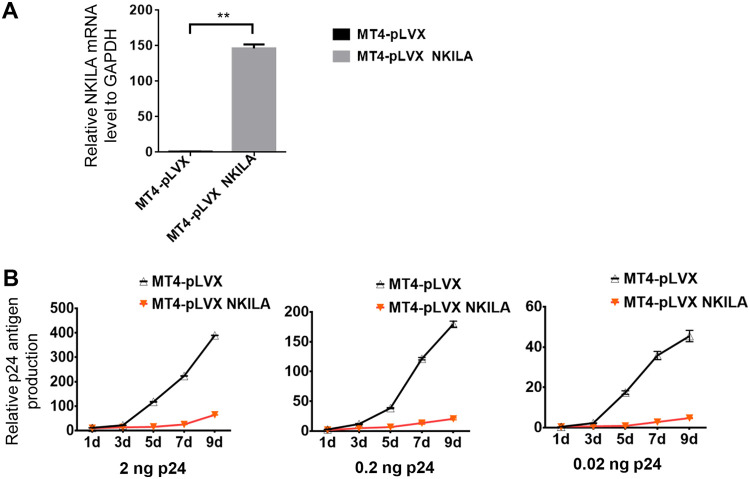
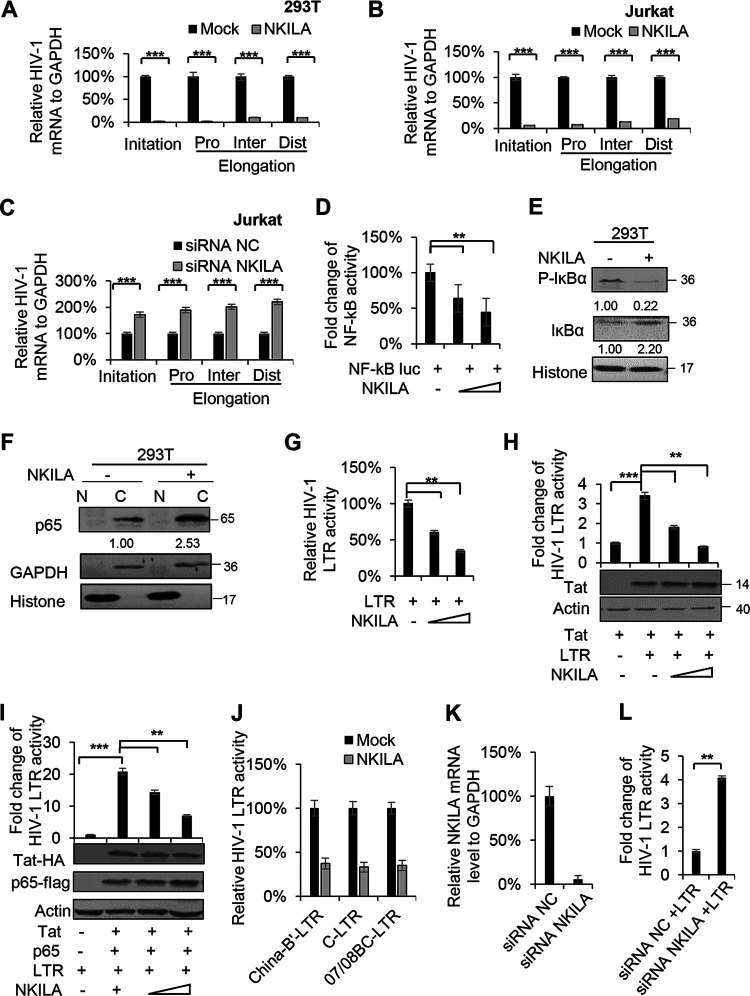
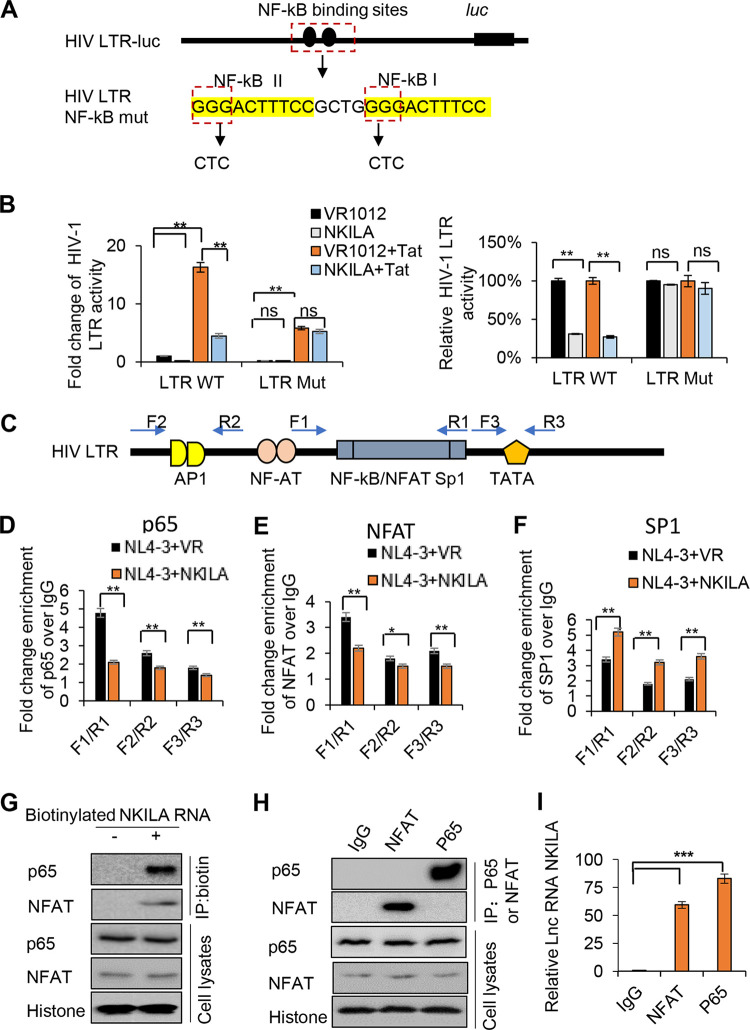
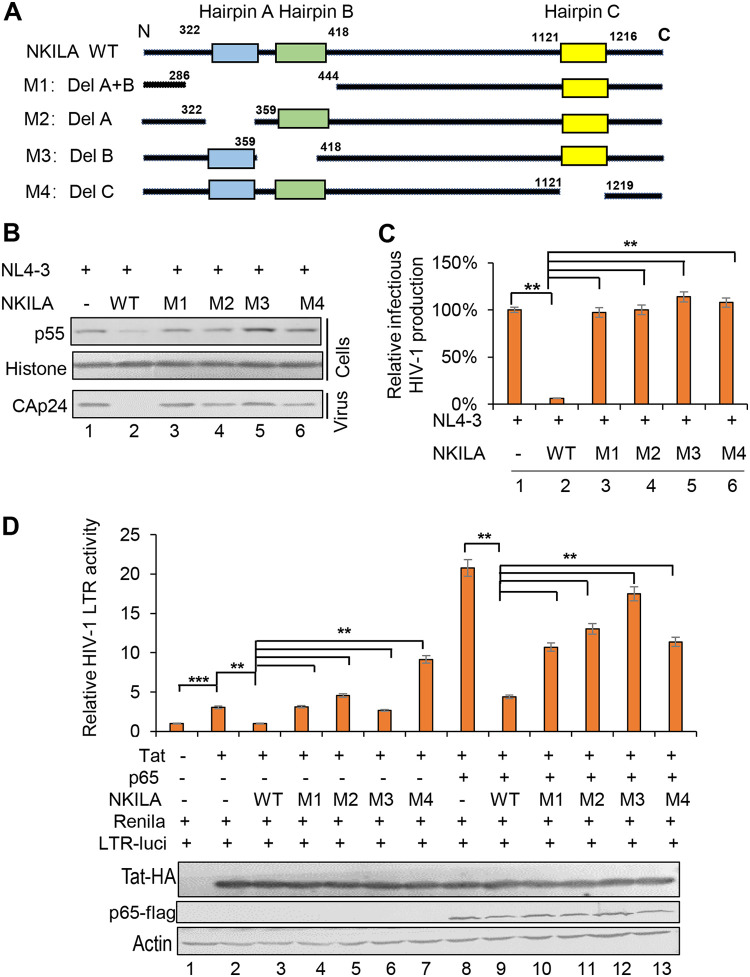
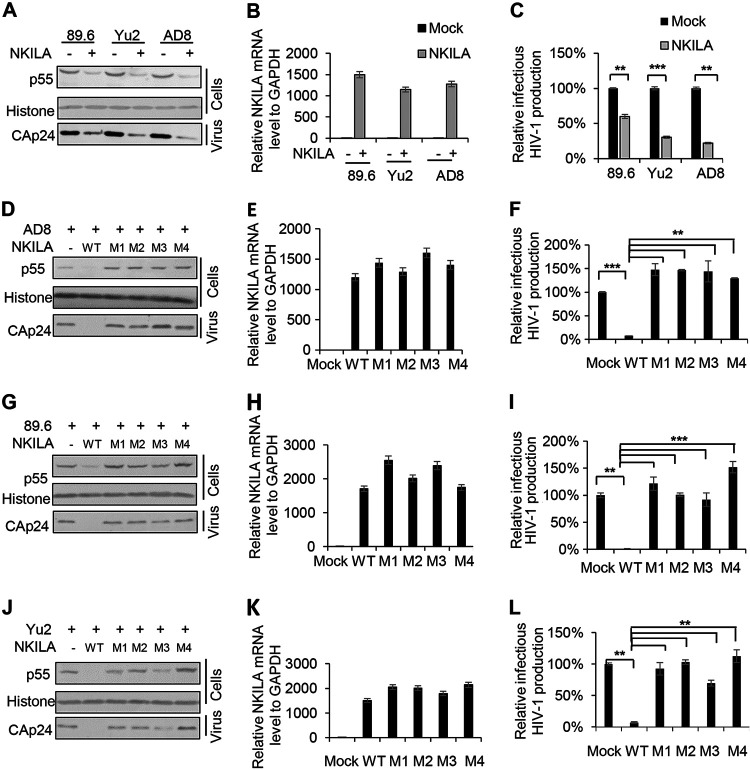
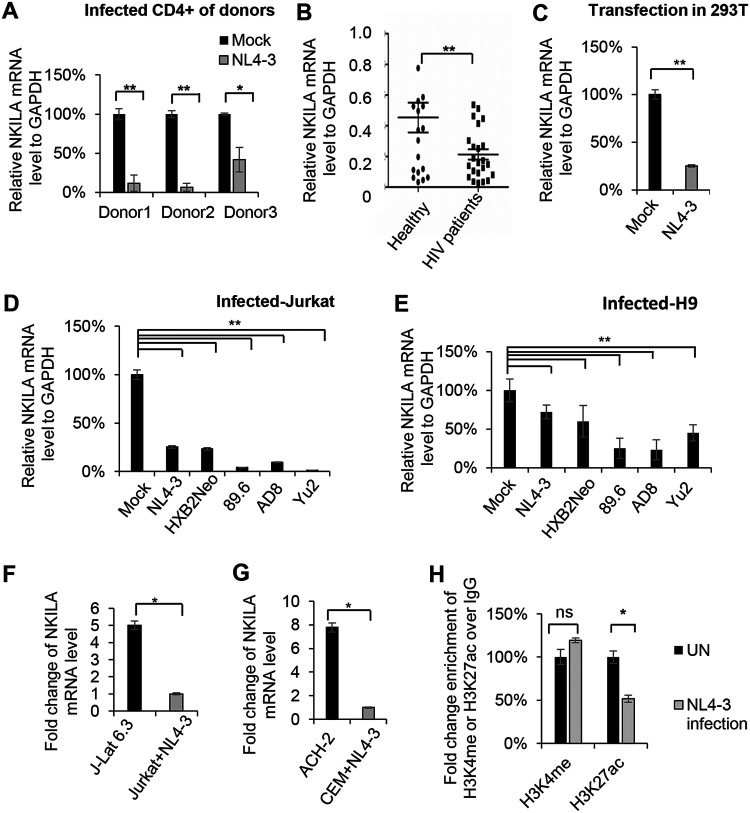
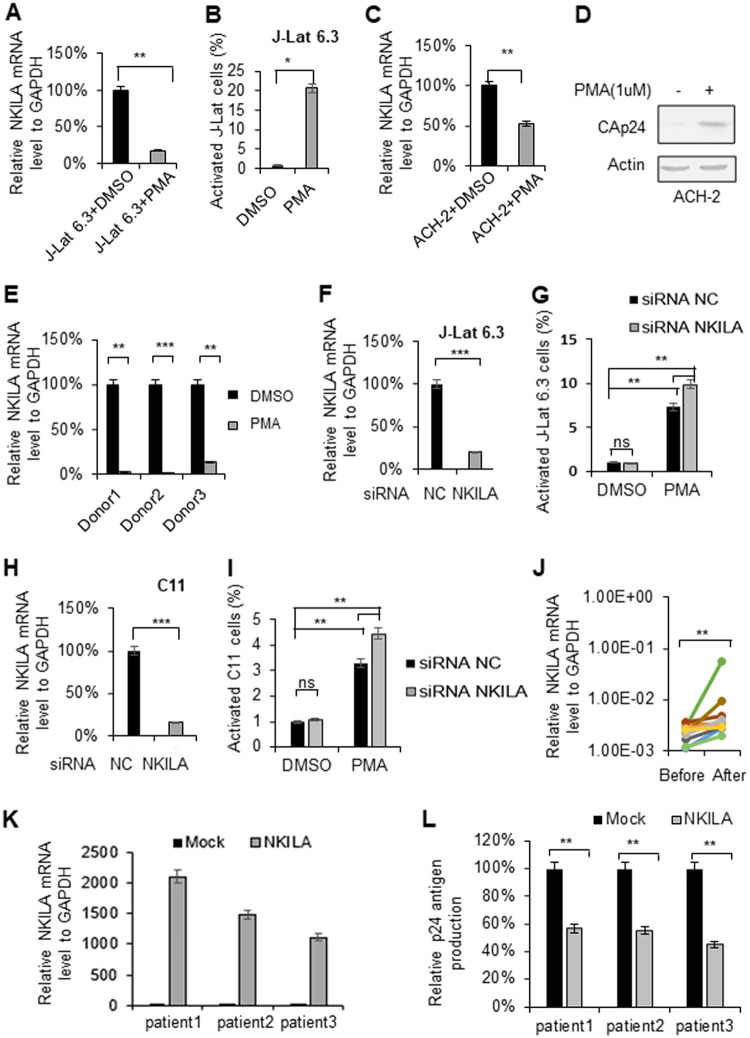
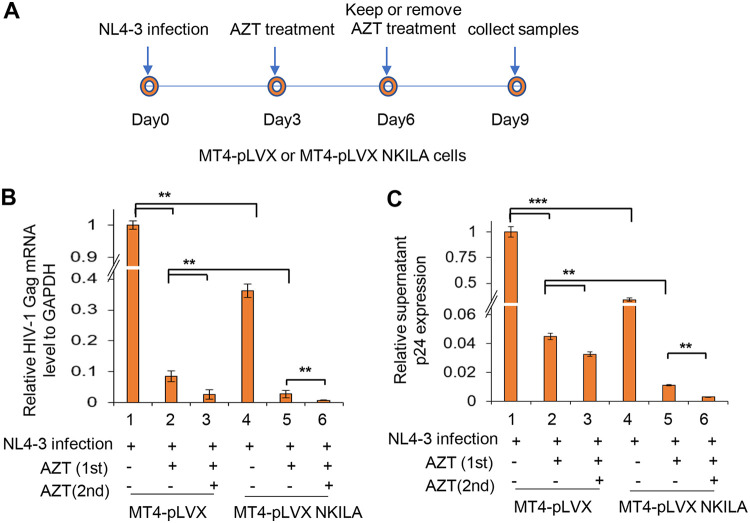
NF-κB-interacting long noncoding RNA (NKILA) was recently identified as a negative regulator of NF-κB signaling and plays an important role in the development of various cancers. It is well known that NF-κB-mediated activation of human immunodeficiency virus type 1 (HIV-1) long terminal repeat (LTR)-driven gene expression is required for HIV-1 transcription and reactivation of latency. However, whether NKILA plays essential roles in HIV-1 replication and latency is unclear. Here, by ectopic expression and silencing experiments, we demonstrate that NKILA potently inhibits HIV-1 replication in an NF-κB-dependent manner by suppressing HIV-1 LTR promoter activity. Moreover, NKILA showed broad-spectrum inhibition on the replication of HIV-1 clones with different coreceptor tropisms as well as on LTR activity of various HIV-1 clinical subtypes. Chromatin immunoprecipitation (ChIP) assays revealed that NKILA expression abolishes the recruitment of p65 to the duplicated κB binding sites in the HIV-1 LTR. NKILA mutants disrupting NF-κB inhibition also lost the ability to inhibit HIV-1 replication. Notably, HIV-1 infection or reactivation significantly downregulated NKILA expression in T cells in order to facilitate viral replication. Downregulated NKILA was mainly due to reduced acetylation of histone K27 on the promoter of NKILA by HIV-1 infection, which blocks NKILA expression. Knockdown of NKILA promoted the reactivation of latent HIV-1 upon phorbol myristate acetate (PMA) stimulation, while ectopic NKILA suppressed the reactivation in a well-established clinical model of withdrawal of azidothymidine (AZT) in vitro These findings improve our understanding of the functional suppression of HIV-1 replication and latency by NKILA through NF-κB signaling.IMPORTANCE The NF-κB pathway plays key roles in HIV-1 replication and reactivation of HIV-1 latency. A regulator inhibiting NF-κB activation may be a promising therapeutic strategy against HIV-1. Recently, NF-κB-interacting long noncoding RNA (NKILA) was identified to suppress the development of different human cancers by inhibiting IκB kinase (IKK)-induced IκB phosphorylation and NF-κB pathway activation, whereas the relationship between NKILA and HIV-1 replication is still unknown. Here, our results show that NKILA inhibits HIV-1 replication and reactivation by suppressing HIV-1 long terminal repeat (LTR)-driven transcription initiation. Moreover, NKILA inhibited the replication of HIV-1 clones with different coreceptor tropisms. This project may reveal a target for the development of novel anti-HIV drugs.
Keywords: HIV-1 latency; HIV-1 replication; NF-κB; NKILA; antiviral activity; lncRNA.
Copyright © 2020 American Society for Microbiology.
Figures
Similar articles
-
Alternate NF-κB-Independent Signaling Reactivation of Latent HIV-1 Provirus.J Virol. 2019 Aug 28;93(18):e00495-19. doi: 10.1128/JVI.00495-19. Print 2019 Sep 15. J Virol. 2019. PMID: 31243131 Free PMC article.
-
Semen Exosomes Promote Transcriptional Silencing of HIV-1 by Disrupting NF-κB/Sp1/Tat Circuitry.J Virol. 2018 Oct 12;92(21):e00731-18. doi: 10.1128/JVI.00731-18. Print 2018 Nov 1. J Virol. 2018. PMID: 30111566 Free PMC article.
-
Naf1 Regulates HIV-1 Latency by Suppressing Viral Promoter-Driven Gene Expression in Primary CD4+ T Cells.J Virol. 2016 Dec 16;91(1):e01830-16. doi: 10.1128/JVI.01830-16. Print 2017 Jan 1. J Virol. 2016. PMID: 27795436 Free PMC article.
-
Efficient Non-Epigenetic Activation of HIV Latency through the T-Cell Receptor Signalosome.Viruses. 2020 Aug 8;12(8):868. doi: 10.3390/v12080868. Viruses. 2020. PMID: 32784426 Free PMC article. Review.
-
NF-κB sub-pathways and HIV cure: A revisit.EBioMedicine. 2021 Jan;63:103159. doi: 10.1016/j.ebiom.2020.103159. Epub 2020 Dec 16. EBioMedicine. 2021. PMID: 33340992 Free PMC article. Review.
Cited by
-
Influence of LncRNA NKILA on Bloodstream Infection of Hypervirulent Klebsiella pneumoniae and Its Ability to Induce Delayed Neutrophil Apoptosis.Evid Based Complement Alternat Med. 2021 Oct 21;2021:6101078. doi: 10.1155/2021/6101078. eCollection 2021. Evid Based Complement Alternat Med. 2021. Retraction in: Evid Based Complement Alternat Med. 2023 Jun 21;2023:9806874. doi: 10.1155/2023/9806874 PMID: 34721638 Free PMC article. Retracted.
-
Non-Coding RNAs in HIV Infection, NeuroHIV, and Related Comorbidities.Cells. 2024 May 23;13(11):898. doi: 10.3390/cells13110898. Cells. 2024. PMID: 38891030 Free PMC article. Review.
-
Breaking the Silence: Regulation of HIV Transcription and Latency on the Road to a Cure.Viruses. 2023 Dec 15;15(12):2435. doi: 10.3390/v15122435. Viruses. 2023. PMID: 38140676 Free PMC article. Review.
-
The Triangle Relationship Between Long Noncoding RNA, RIG-I-like Receptor Signaling Pathway, and Glycolysis.Front Microbiol. 2021 Nov 30;12:807737. doi: 10.3389/fmicb.2021.807737. eCollection 2021. Front Microbiol. 2021. PMID: 34917069 Free PMC article. Review.
-
Unveiling the Hidden Regulators: The Impact of lncRNAs on Zoonoses.Int J Mol Sci. 2024 Mar 21;25(6):3539. doi: 10.3390/ijms25063539. Int J Mol Sci. 2024. PMID: 38542512 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
