

Rampant C→U Hypermutation in the Genomes of SARS-CoV-2 and Other Coronaviruses: Causes and Consequences for Their Short- and Long-Term Evolutionary Trajectories
- PMID: 32581081
- PMCID: PMC7316492
- DOI: 10.1128/mSphere.00408-20
Rampant C→U Hypermutation in the Genomes of SARS-CoV-2 and Other Coronaviruses: Causes and Consequences for Their Short- and Long-Term Evolutionary Trajectories
Abstract
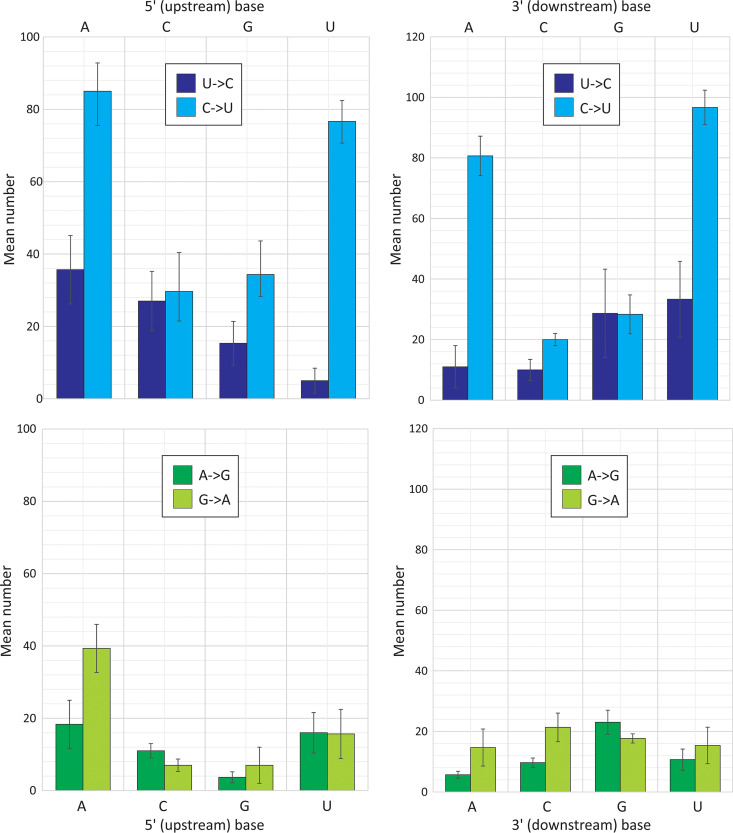
The pandemic of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has motivated an intensive analysis of its molecular epidemiology following its worldwide spread. To understand the early evolutionary events following its emergence, a data set of 985 complete SARS-CoV-2 sequences was assembled. Variants showed a mean of 5.5 to 9.5 nucleotide differences from each other, consistent with a midrange coronavirus substitution rate of 3 × 10-4 substitutions/site/year. Almost one-half of sequence changes were C→U transitions, with an 8-fold base frequency normalized directional asymmetry between C→U and U→C substitutions. Elevated ratios were observed in other recently emerged coronaviruses (SARS-CoV, Middle East respiratory syndrome [MERS]-CoV), and decreasing ratios were observed in other human coronaviruses (HCoV-NL63, -OC43, -229E, and -HKU1) proportionate to their increasing divergence. C→U transitions underpinned almost one-half of the amino acid differences between SARS-CoV-2 variants and occurred preferentially in both 5' U/A and 3' U/A flanking sequence contexts comparable to favored motifs of human APOBEC3 proteins. Marked base asymmetries observed in nonpandemic human coronaviruses (U ≫ A > G ≫ C) and low G+C contents may represent long-term effects of prolonged C→U hypermutation in their hosts. The evidence that much of sequence change in SARS-CoV-2 and other coronaviruses may be driven by a host APOBEC-like editing process has profound implications for understanding their short- and long-term evolution. Repeated cycles of mutation and reversion in favored mutational hot spots and the widespread occurrence of amino acid changes with no adaptive value for the virus represent a quite different paradigm of virus sequence change from neutral and Darwinian evolutionary frameworks and are not incorporated by standard models used in molecular epidemiology investigations.IMPORTANCE The wealth of accurately curated sequence data for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), its long genome, and its low substitution rate provides a relatively blank canvas with which to investigate effects of mutational and editing processes imposed by the host cell. The finding that a large proportion of sequence change in SARS-CoV-2 in the initial months of the pandemic comprised C→U mutations in a host APOBEC-like context provides evidence for a potent host-driven antiviral editing mechanism against coronaviruses more often associated with antiretroviral defense. In evolutionary terms, the contribution of biased, convergent, and context-dependent mutations to sequence change in SARS-CoV-2 is substantial, and these processes are not incorporated by standard models used in molecular epidemiology investigations.
Keywords: APOBEC; COVID-19; SARS; SARS coronavirus 2; SARS-CoV-2; coronavirus; hypermutation.
Copyright © 2020 Simmonds.
Figures






Similar articles
-
Coronavirus genomes carry the signatures of their habitats.PLoS One. 2020 Dec 22;15(12):e0244025. doi: 10.1371/journal.pone.0244025. eCollection 2020. PLoS One. 2020. PMID: 33351847 Free PMC article.
-
Ancestral origin, antigenic resemblance and epidemiological insights of novel coronavirus (SARS-CoV-2): Global burden and Bangladesh perspective.Infect Genet Evol. 2020 Oct;84:104440. doi: 10.1016/j.meegid.2020.104440. Epub 2020 Jul 1. Infect Genet Evol. 2020. PMID: 32622082 Free PMC article.
-
Potential APOBEC-mediated RNA editing of the genomes of SARS-CoV-2 and other coronaviruses and its impact on their longer term evolution.Virology. 2021 Apr;556:62-72. doi: 10.1016/j.virol.2020.12.018. Epub 2021 Jan 7. Virology. 2021. PMID: 33545556 Free PMC article. Review.
-
Mutation Patterns of Human SARS-CoV-2 and Bat RaTG13 Coronavirus Genomes Are Strongly Biased Towards C>U Transitions, Indicating Rapid Evolution in Their Hosts.Genes (Basel). 2020 Jul 7;11(7):761. doi: 10.3390/genes11070761. Genes (Basel). 2020. PMID: 32646049 Free PMC article.
-
Coronaviruses and SARS-COV-2.Turk J Med Sci. 2020 Apr 21;50(SI-1):549-556. doi: 10.3906/sag-2004-127. Turk J Med Sci. 2020. PMID: 32293832 Free PMC article. Review.
Cited by
-
Impact of Vaccination on Intra-Host Genetic Diversity of Patients Infected with SARS-CoV-2 Gamma Lineage.Viruses. 2024 Sep 26;16(10):1524. doi: 10.3390/v16101524. Viruses. 2024. PMID: 39459859 Free PMC article.
-
Host-dependent C-to-U RNA editing in SARS-CoV-2 creates novel viral genes with optimized expressibility.Front Cell Infect Microbiol. 2024 Oct 9;14:1476605. doi: 10.3389/fcimb.2024.1476605. eCollection 2024. Front Cell Infect Microbiol. 2024. PMID: 39445213 Free PMC article.
-
COVID-19 mutations: An overview.World J Methodol. 2024 Sep 20;14(3):89761. doi: 10.5662/wjm.v14.i3.89761. eCollection 2024 Sep 20. World J Methodol. 2024. PMID: 39310238 Free PMC article. Review.
-
Prediction of the effects of the top 10 synonymous mutations from 26645 SARS-CoV-2 genomes of early pandemic phase.F1000Res. 2024 Sep 18;10:1053. doi: 10.12688/f1000research.72896.3. eCollection 2021. F1000Res. 2024. PMID: 39268187 Free PMC article.
-
Potential Role of APOBEC3 Family Proteins in SARS-CoV-2 Replication.Viruses. 2024 Jul 16;16(7):1141. doi: 10.3390/v16071141. Viruses. 2024. PMID: 39066304 Free PMC article.
References
-
- Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, Ren R, Leung KSM, Lau EHY, Wong JY, Xing X, Xiang N, Wu Y, Li C, Chen Q, Li D, Liu T, Zhao J, Liu M, Tu W, Chen C, Jin L, Yang R, Wang Q, Zhou S, Wang R, Liu H, Luo Y, Liu Y, Shao G, Li H, Tao Z, Yang Y, Deng Z, Liu B, Ma Z, Zhang Y, Shi G, Lam TTY, Wu JT, Gao GF, Cowling BJ, Yang B, Leung GM, Feng Z. 2020. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med 382:1199–1207. doi:10.1056/NEJMoa2001316. - DOI - PMC - PubMed
-
- Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. doi:10.1038/s41586-020-2012-7. - DOI - PMC - PubMed
-
- Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W, China Novel Coronavirus Investigating and Research Team. 2020. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727–733. doi:10.1056/NEJMoa2001017. - DOI - PMC - PubMed
-
- Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, Zhang YZ. 2020. A new coronavirus associated with human respiratory disease in China. Nature 579:265–269. doi:10.1038/s41586-020-2008-3. - DOI - PMC - PubMed
-
- Mavian C, Marini S, Manes C, Capua I, Prosperi M, Salemi M. 20 March 2020. Regaining perspective on SARS-CoV-2 molecular tracing and its implications. medRxiv https://www.medrxiv.org/content/10.1101/2020.03.16.20034470v1. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
