

Altered RNA Splicing by Mutant p53 Activates Oncogenic RAS Signaling in Pancreatic Cancer
- PMID: 32559497
- PMCID: PMC8028848
- DOI: 10.1016/j.ccell.2020.05.010
Altered RNA Splicing by Mutant p53 Activates Oncogenic RAS Signaling in Pancreatic Cancer
Abstract
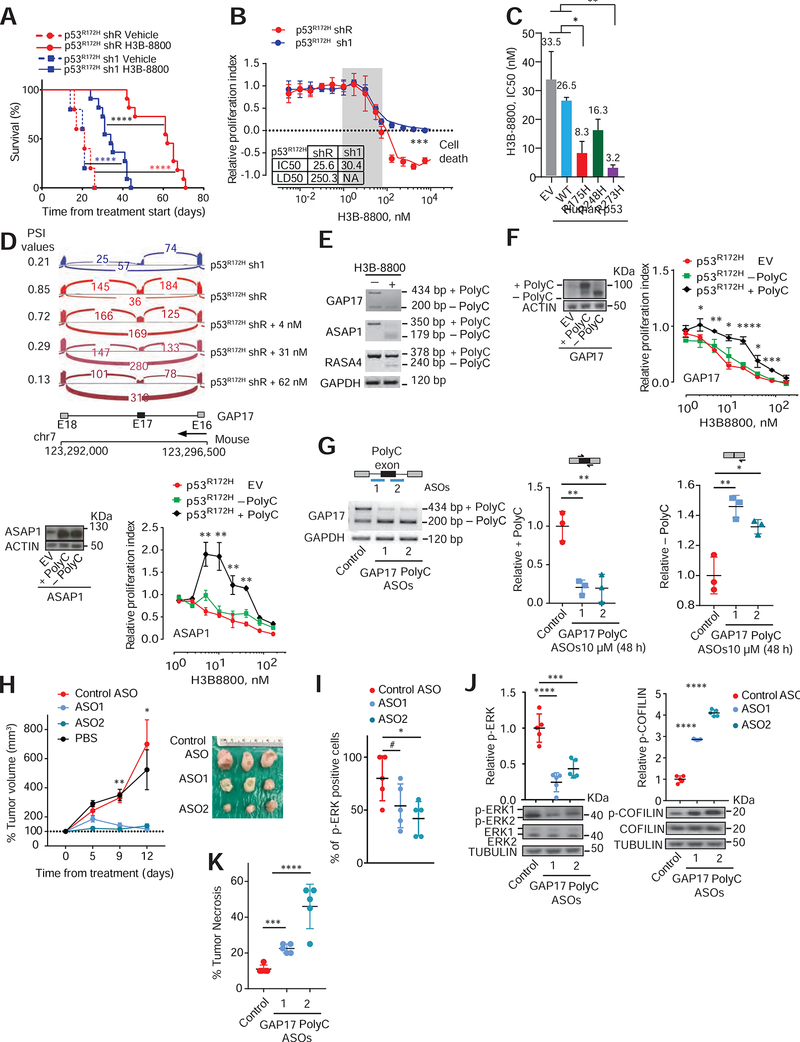
Pancreatic ductal adenocarcinoma (PDAC) is driven by co-existing mutations in KRAS and TP53. However, how these mutations collaborate to promote this cancer is unknown. Here, we uncover sequence-specific changes in RNA splicing enforced by mutant p53 which enhance KRAS activity. Mutant p53 increases expression of splicing regulator hnRNPK to promote inclusion of cytosine-rich exons within GTPase-activating proteins (GAPs), negative regulators of RAS family members. Mutant p53-enforced GAP isoforms lose cell membrane association, leading to heightened KRAS activity. Preventing cytosine-rich exon inclusion in mutant KRAS/p53 PDACs decreases tumor growth. Moreover, mutant p53 PDACs are sensitized to inhibition of splicing via spliceosome inhibitors. These data provide insight into co-enrichment of KRAS and p53 mutations and therapeutics targeting this mechanism in PDAC.
Keywords: GAP17; GTPase signaling; KRAS; RNA splicing; SF3B1; hnRNPK; oncogenes; p53; pancreatic cancer; splicing inhibitors.
Copyright © 2020. Published by Elsevier Inc.
Conflict of interest statement
Declaration of Interests L.F.E.-H. is a consultant for KDx Diagnostics Inc and OncoGenesis Inc. O.A.-W. served as a consultant for H3B Biomedicine, Foundation Medicine Inc, Merck, and Janssen, and is on the scientific advisory board of Envisagenics; O.A.-W. received prior research funding from H3B Biomedicine unrelated to the current manuscript and serves on the Scientific Advisory Board of Envisagenics Inc. S.D.L. is a member of the Scientific Advisory Board for Nybo Pharmaceuticals and co-founder of Episteme Prognostics.
Figures
Similar articles
-
Oncogenic KRAS Reduces Expression of FGF21 in Acinar Cells to Promote Pancreatic Tumorigenesis in Mice on a High-Fat Diet.Gastroenterology. 2019 Nov;157(5):1413-1428.e11. doi: 10.1053/j.gastro.2019.07.030. Epub 2019 Jul 25. Gastroenterology. 2019. PMID: 31352001 Free PMC article.
-
Nicotine promotes initiation and progression of KRAS-induced pancreatic cancer via Gata6-dependent dedifferentiation of acinar cells in mice.Gastroenterology. 2014 Nov;147(5):1119-33.e4. doi: 10.1053/j.gastro.2014.08.002. Epub 2014 Aug 12. Gastroenterology. 2014. PMID: 25127677
-
Oncogenic ERBB2 aberrations and KRAS mutations cooperate to promote pancreatic ductal adenocarcinoma progression.Carcinogenesis. 2020 Mar 13;41(1):44-55. doi: 10.1093/carcin/bgz086. Carcinogenesis. 2020. PMID: 31046123
-
Highlights on the Role of KRAS Mutations in Reshaping the Microenvironment of Pancreatic Adenocarcinoma.Int J Mol Sci. 2021 Sep 23;22(19):10219. doi: 10.3390/ijms221910219. Int J Mol Sci. 2021. PMID: 34638560 Free PMC article. Review.
-
Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: Hopes and realities.Eur J Cancer. 2016 Feb;54:75-83. doi: 10.1016/j.ejca.2015.11.012. Epub 2015 Dec 28. Eur J Cancer. 2016. PMID: 26735353 Review.
Cited by
-
Mutant p53 in cell-cell interactions.Genes Dev. 2021 Apr 1;35(7-8):433-448. doi: 10.1101/gad.347542.120. Genes Dev. 2021. PMID: 33861719 Free PMC article. Review.
-
Mutant p53 gain of function mediates cancer immune escape that is counteracted by APR-246.Br J Cancer. 2022 Nov;127(11):2060-2071. doi: 10.1038/s41416-022-01971-8. Epub 2022 Sep 22. Br J Cancer. 2022. PMID: 36138076 Free PMC article.
-
Identification of LncRNA Prognostic Signature Associated With Genomic Instability in Pancreatic Adenocarcinoma.Front Oncol. 2022 Mar 29;12:799475. doi: 10.3389/fonc.2022.799475. eCollection 2022. Front Oncol. 2022. PMID: 35433487 Free PMC article.
-
Clinical Impact of Molecular Subtyping of Pancreatic Cancer.Front Cell Dev Biol. 2021 Nov 5;9:743908. doi: 10.3389/fcell.2021.743908. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34805152 Free PMC article. Review.
-
Steering research on mRNA splicing in cancer towards clinical translation.Nat Rev Cancer. 2024 Dec;24(12):887-905. doi: 10.1038/s41568-024-00750-2. Epub 2024 Oct 9. Nat Rev Cancer. 2024. PMID: 39384951 Free PMC article. Review.
References
-
- Agbunag C, Lee KE, Buontempo S, and Bar-Sagi D (2006). Pancreatic duct epithelial cell isolation and cultivation in two-dimensional and three-dimensional culture systems. Methods Enzymol 407, 703–710. - PubMed
-
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
