

Mitochondrial AIF loss causes metabolic reprogramming, caspase-independent cell death blockade, embryonic lethality, and perinatal hydrocephalus
- PMID: 32480041
- PMCID: PMC7334469
- DOI: 10.1016/j.molmet.2020.101027
Mitochondrial AIF loss causes metabolic reprogramming, caspase-independent cell death blockade, embryonic lethality, and perinatal hydrocephalus
Abstract
Objectives: Apoptosis-Inducing Factor (AIF) is a protein involved in mitochondrial electron transport chain assembly/stability and programmed cell death. The relevant role of this protein is underlined because mutations altering mitochondrial AIF properties result in acute pediatric mitochondriopathies and tumor metastasis. By generating an original AIF-deficient mouse strain, this study attempted to analyze, in a single paradigm, the cellular and developmental metabolic consequences of AIF loss and the subsequent oxidative phosphorylation (OXPHOS) dysfunction.
Methods: We developed a novel AIF-deficient mouse strain and assessed, using molecular and cell biology approaches, the cellular, embryonic, and adult mice phenotypic alterations. Additionally, we conducted ex vivo assays with primary and immortalized AIF knockout mouse embryonic fibroblasts (MEFs) to establish the cell death characteristics and the metabolic adaptive responses provoked by the mitochondrial electron transport chain (ETC) breakdown.
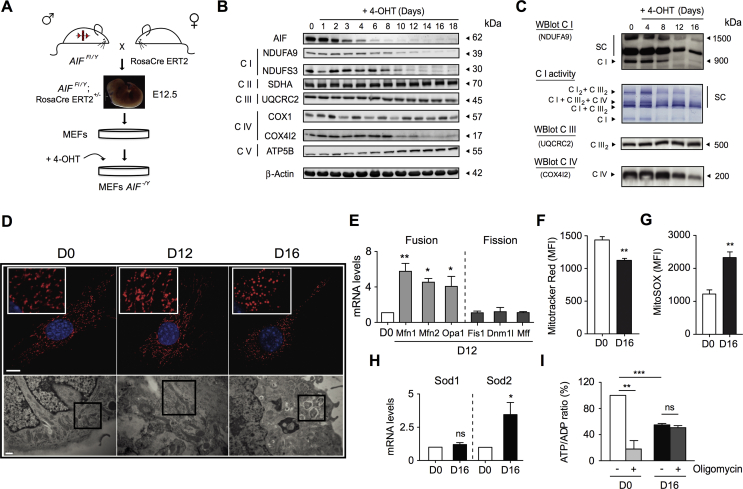
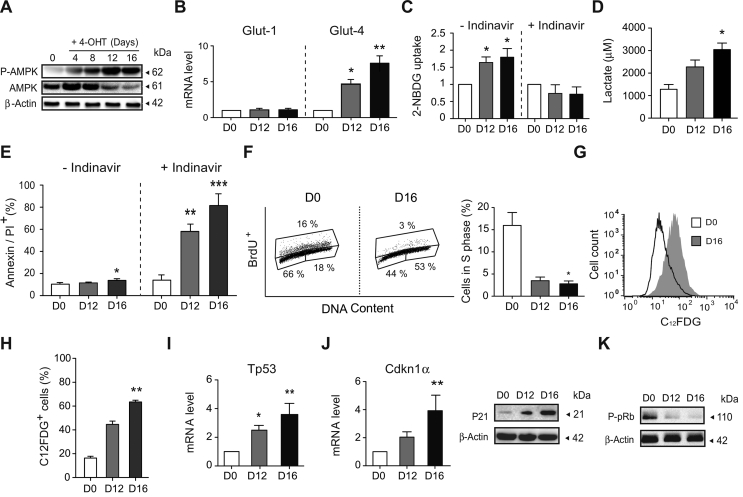
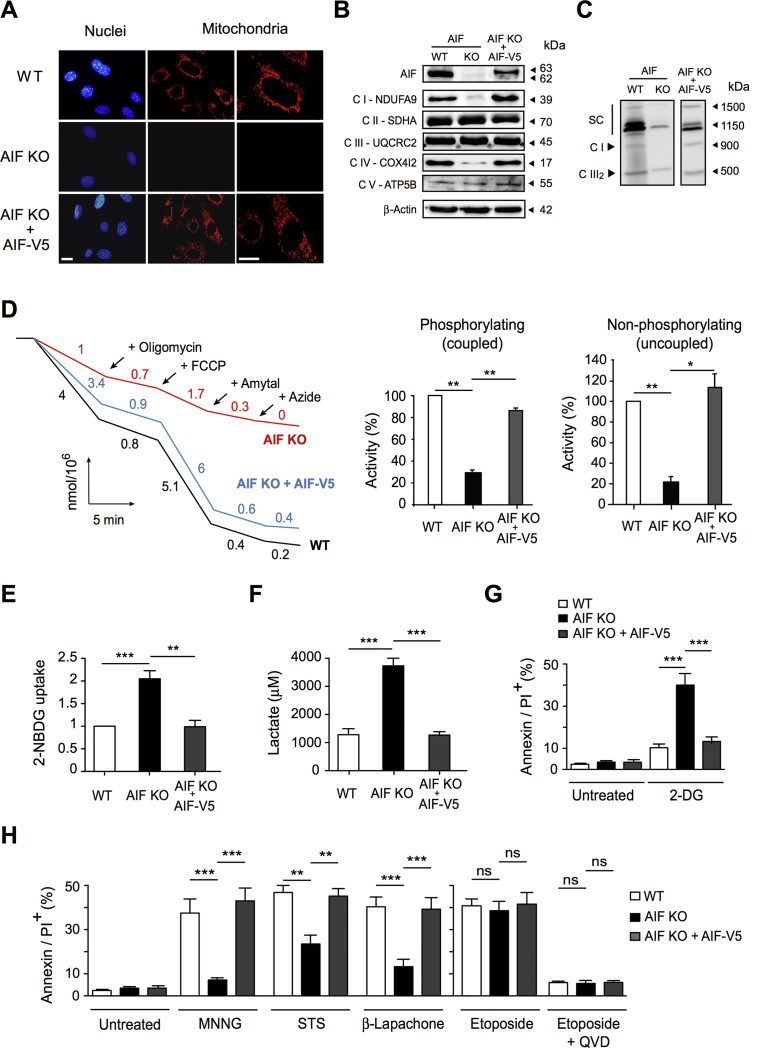
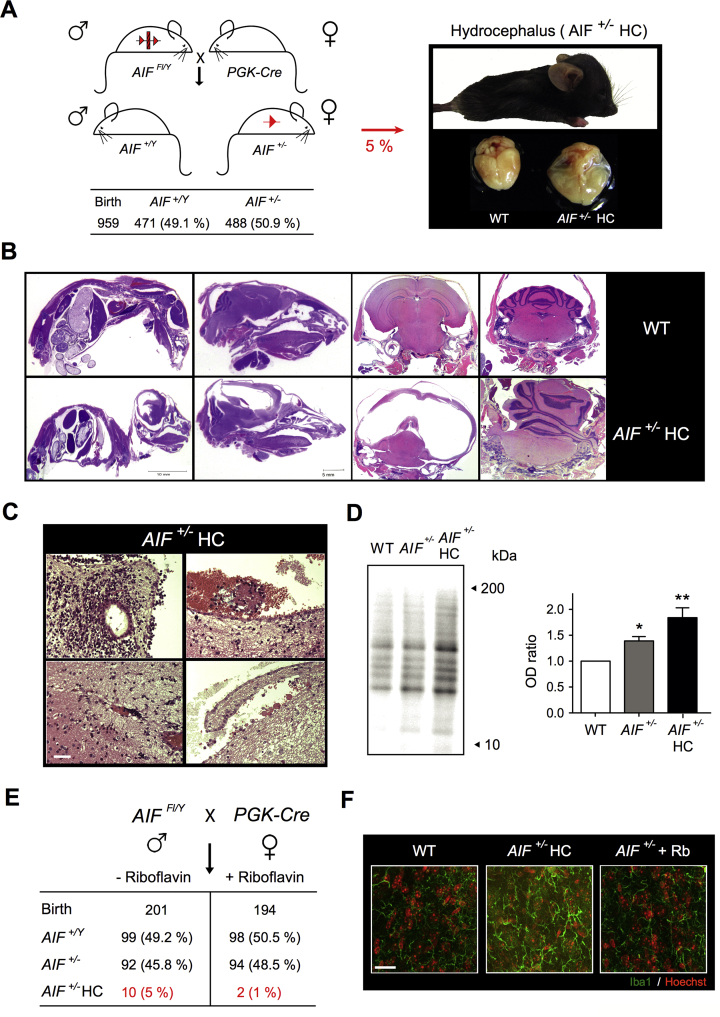
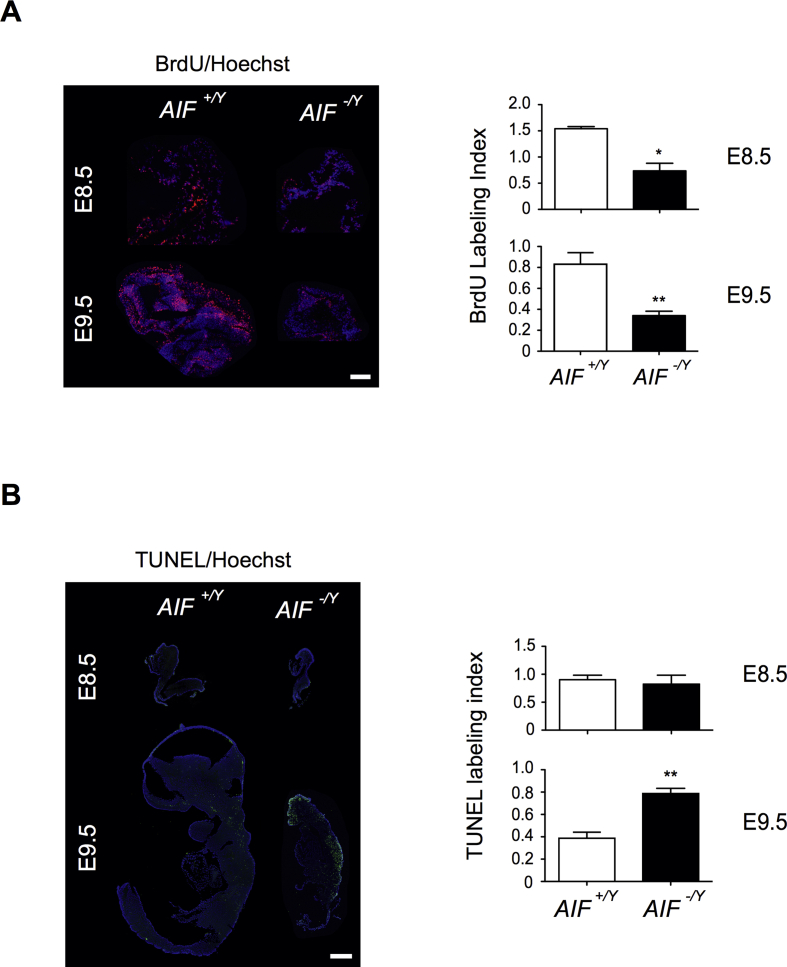
Results: AIF deficiency destabilized mitochondrial ETC and provoked supercomplex disorganization, mitochondrial transmembrane potential loss, and high generation of mitochondrial reactive oxygen species (ROS). AIF-/Y MEFs counterbalanced these OXPHOS alterations by mitochondrial network reorganization and a metabolic reprogramming toward anaerobic glycolysis illustrated by the AMPK phosphorylation at Thr172, the overexpression of the glucose transporter GLUT-4, the subsequent enhancement of glucose uptake, and the anaerobic lactate generation. A late phenotype was characterized by the activation of P53/P21-mediated senescence. Notably, approximately 2% of AIF-/Y MEFs diminished both mitochondrial mass and ROS levels and spontaneously proliferated. These cycling AIF-/Y MEFs were resistant to caspase-independent cell death inducers. The AIF-deficient mouse strain was embryonic lethal between E11.5 and E13.5 with energy loss, proliferation arrest, and increased apoptotic levels. Contrary to AIF-/Y MEFs, the AIF KO embryos were unable to reprogram their metabolism toward anaerobic glycolysis. Heterozygous AIF+/- females displayed progressive bone marrow, thymus, and spleen cellular loss. In addition, approximately 10% of AIF+/- females developed perinatal hydrocephaly characterized by brain development impairment, meningeal fibrosis, and medullar hemorrhages; those mice died 5 weeks after birth. AIF+/- with hydrocephaly exhibited loss of ciliated epithelium in the ependymal layer. This phenotype was triggered by the ROS excess. Accordingly, it was possible to diminish the occurrence of hydrocephalus AIF+/- females by supplying dams and newborns with an antioxidant in drinking water.
Conclusions: In a single knockout model and at 3 different levels (cell, embryo, and adult mice) we demonstrated that by controlling the mitochondrial OXPHOS/metabolism, AIF is a key factor regulating cell differentiation and fate. Additionally, by providing new insights into the pathological consequences of mitochondrial OXPHOS dysfunction, our new findings pave the way for novel pharmacological strategies.
Keywords: AIF; Caspase-independent cell death; Hydrocephaly; Metabolism; Mitochondria; OXPHOS.
Copyright © 2020 The Author(s). Published by Elsevier GmbH.. All rights reserved.
Figures

Similar articles
-
AIF loss deregulates hematopoiesis and reveals different adaptive metabolic responses in bone marrow cells and thymocytes.Cell Death Differ. 2018 May;25(5):983-1001. doi: 10.1038/s41418-017-0035-x. Epub 2018 Jan 11. Cell Death Differ. 2018. PMID: 29323266 Free PMC article.
-
Deficiency in Apoptosis-Inducing Factor Recapitulates Chronic Kidney Disease via Aberrant Mitochondrial Homeostasis.Diabetes. 2016 Apr;65(4):1085-98. doi: 10.2337/db15-0864. Epub 2016 Jan 28. Diabetes. 2016. PMID: 26822084
-
Potential role of Apoptosis Inducing Factor in evolutionarily significant eukaryote, Dictyostelium discoideum survival.Biochim Biophys Acta Gen Subj. 2017 Jan;1861(1 Pt A):2942-2955. doi: 10.1016/j.bbagen.2016.09.021. Epub 2016 Sep 20. Biochim Biophys Acta Gen Subj. 2017. PMID: 27663234
-
Apoptosis-Inducing Factor (AIF) in Physiology and Disease: The Tale of a Repented Natural Born Killer.EBioMedicine. 2018 Apr;30:29-37. doi: 10.1016/j.ebiom.2018.03.016. Epub 2018 Mar 23. EBioMedicine. 2018. PMID: 29605508 Free PMC article. Review.
-
Role of apoptosis-inducing factor (Aif) in the T cell lineage.Indian J Med Res. 2013 Nov;138(5):577-90. Indian J Med Res. 2013. PMID: 24434313 Free PMC article. Review.
Cited by
-
Hacd2 deficiency in mice leads to an early and lethal mitochondrial disease.Mol Metab. 2023 Mar;69:101677. doi: 10.1016/j.molmet.2023.101677. Epub 2023 Jan 21. Mol Metab. 2023. PMID: 36693621 Free PMC article.
-
PHB2 promotes colorectal cancer cell proliferation and tumorigenesis through NDUFS1-mediated oxidative phosphorylation.Cell Death Dis. 2023 Jan 20;14(1):44. doi: 10.1038/s41419-023-05575-9. Cell Death Dis. 2023. PMID: 36658121 Free PMC article.
-
W196 and the β-Hairpin Motif Modulate the Redox Switch of Conformation and the Biomolecular Interaction Network of the Apoptosis-Inducing Factor.Oxid Med Cell Longev. 2021 Jan 15;2021:6673661. doi: 10.1155/2021/6673661. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 33510840 Free PMC article.
-
Beyond a platform protein for the degradosome assembly: The Apoptosis-Inducing Factor as an efficient nuclease involved in chromatinolysis.PNAS Nexus. 2022 Dec 26;2(2):pgac312. doi: 10.1093/pnasnexus/pgac312. eCollection 2023 Feb. PNAS Nexus. 2022. PMID: 36845352 Free PMC article.
-
Reactive microglia and mitochondrial unfolded protein response following ventriculomegaly and behavior defects in kaolin-induced hydrocephalus.BMB Rep. 2022 Apr;55(4):181-186. doi: 10.5483/BMBRep.2022.55.4.126. BMB Rep. 2022. PMID: 34903317 Free PMC article.
References
-
- Pearce E.L., Poffenberger M.C., Chang C.H., Jones R.G. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342(6155):1242454. - PubMed
-
- Holmstrom K.M., Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nature Reviews Molecular Cell Biology. 2014;15(6):411–421. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
