

Prion protein post-translational modifications modulate heparan sulfate binding and limit aggregate size in prion disease
- PMID: 32454127
- PMCID: PMC7425694
- DOI: 10.1016/j.nbd.2020.104955
Prion protein post-translational modifications modulate heparan sulfate binding and limit aggregate size in prion disease
Abstract
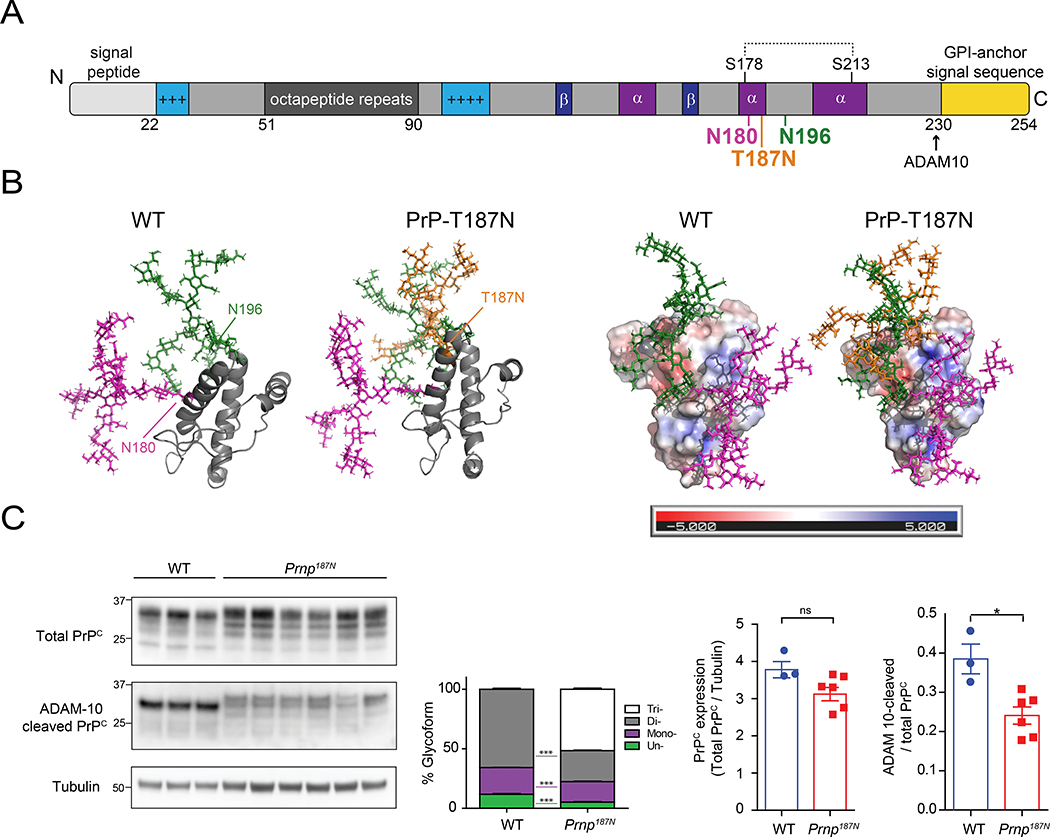
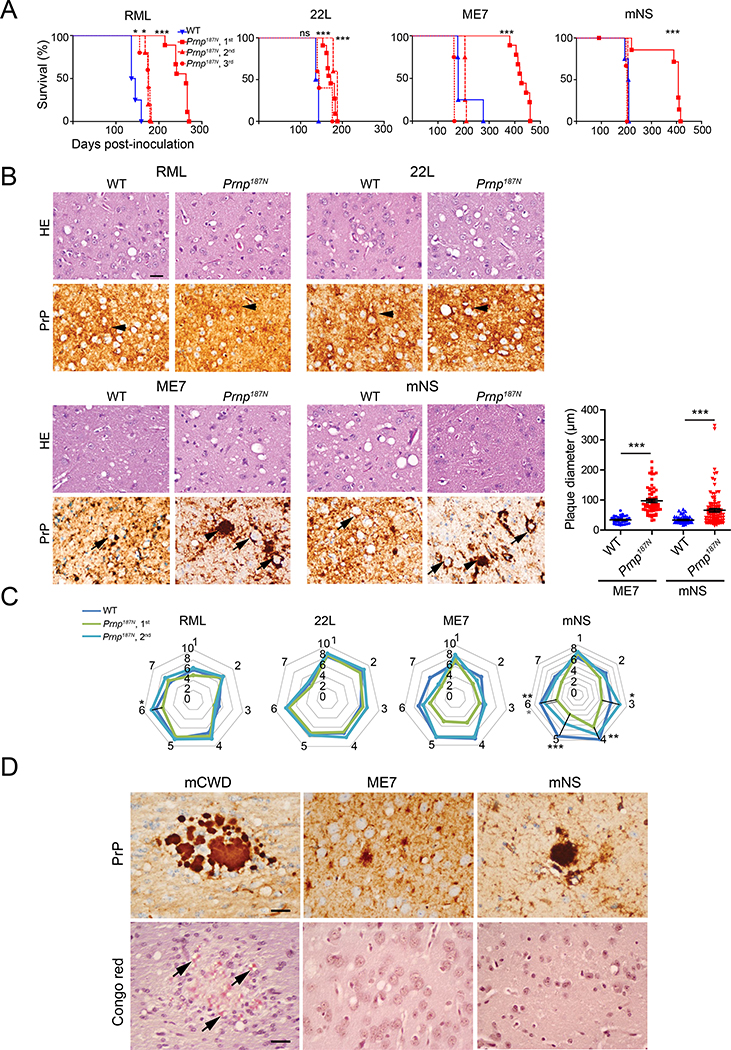
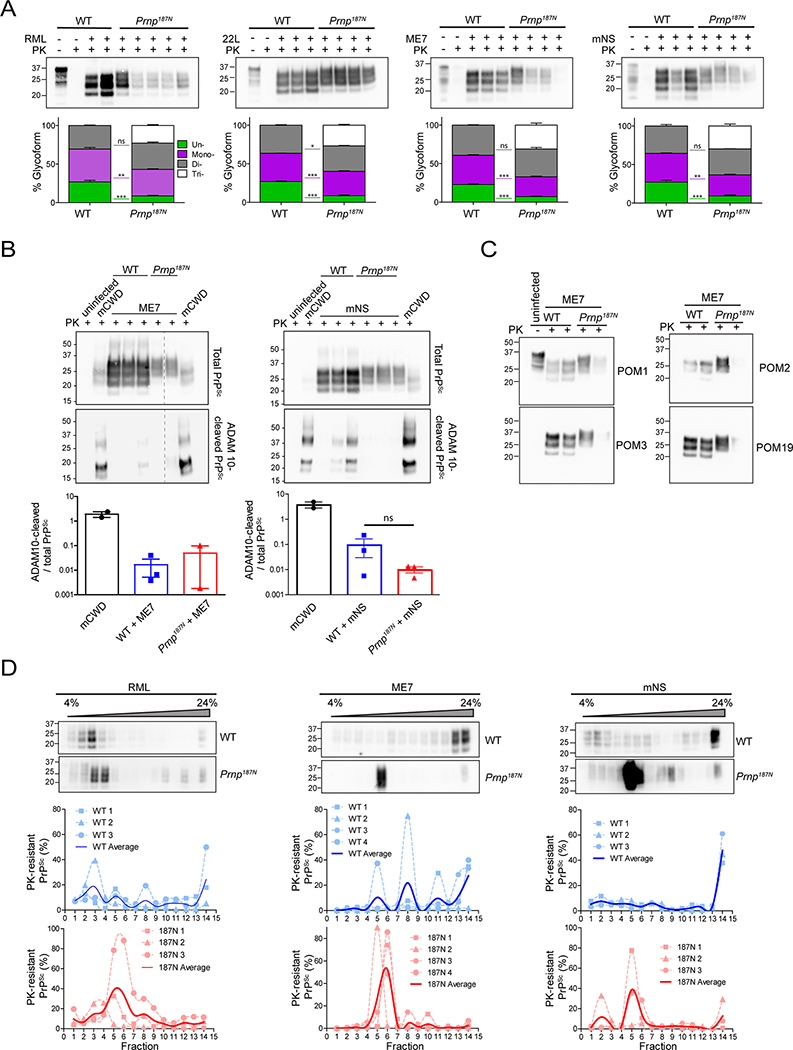
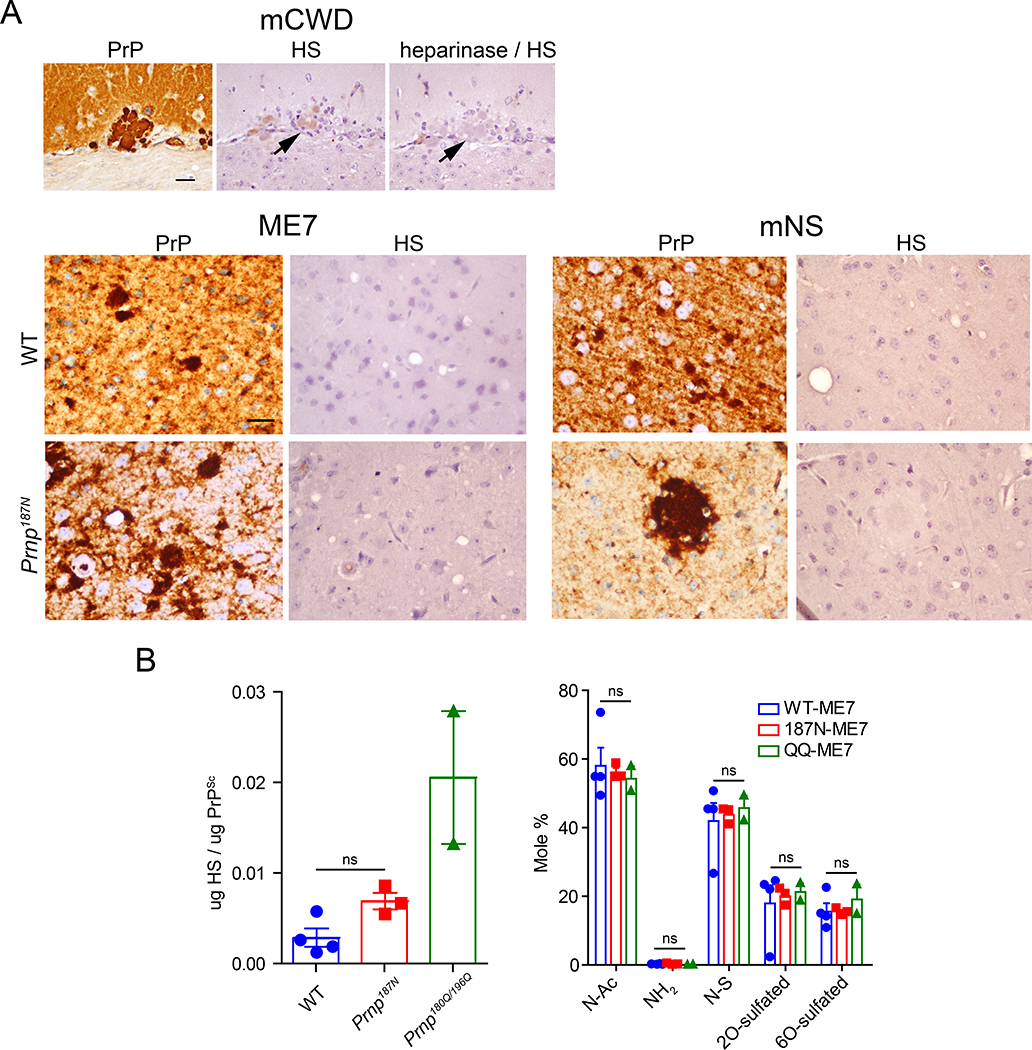
Many aggregation-prone proteins linked to neurodegenerative disease are post-translationally modified during their biogenesis. In vivo pathogenesis studies have suggested that the presence of post-translational modifications can shift the aggregate assembly pathway and profoundly alter the disease phenotype. In prion disease, the N-linked glycans and GPI-anchor on the prion protein (PrP) impair fibril assembly. However, the relevance of the two glycans to aggregate structure and disease progression remains unclear. Here we show that prion-infected knockin mice expressing an additional PrP glycan (tri-glycosylated PrP) develop new plaque-like deposits on neuronal cell membranes, along the subarachnoid space, and periventricularly, suggestive of high prion mobility and transit through the interstitial fluid. These plaque-like deposits were largely non-congophilic and composed of full length, uncleaved PrP, indicating retention of the glycophosphatidylinositol (GPI) anchor. Prion aggregates sedimented in low density fractions following ultracentrifugation, consistent with oligomers, and bound low levels of heparan sulfate (HS) similar to other predominantly GPI-anchored prions. Collectively, these results suggest that highly glycosylated PrP primarily converts as a GPI-anchored glycoform, with low involvement of HS co-factors, limiting PrP assembly mainly to oligomers. Since PrPC is highly glycosylated, these findings may explain the high frequency of diffuse, synaptic, and plaque-like deposits in the brain as well as the rapid conversion commonly observed in human and animal prion disease.
Keywords: ADAM10 cleavage; Amyloid; Glycans; Glycosaminoglycans; Glycosylation; Neurodegeneration; Prion strains; Protein misfolding.
Copyright © 2020. Published by Elsevier Inc.
Conflict of interest statement
Declaration of Competing Interest
The authors declare no competing financial interests.
Figures
Similar articles
-
Prion protein glycans reduce intracerebral fibril formation and spongiosis in prion disease.J Clin Invest. 2020 Mar 2;130(3):1350-1362. doi: 10.1172/JCI131564. J Clin Invest. 2020. PMID: 31985492 Free PMC article.
-
Prion-associated cerebral amyloid angiopathy is not exacerbated by human phosphorylated tau aggregates in scrapie-infected mice expressing anchorless prion protein.Neurobiol Dis. 2020 Oct;144:105057. doi: 10.1016/j.nbd.2020.105057. Epub 2020 Aug 21. Neurobiol Dis. 2020. PMID: 32829029
-
Shortening heparan sulfate chains prolongs survival and reduces parenchymal plaques in prion disease caused by mobile, ADAM10-cleaved prions.Acta Neuropathol. 2020 Mar;139(3):527-546. doi: 10.1007/s00401-019-02085-x. Epub 2019 Oct 31. Acta Neuropathol. 2020. PMID: 31673874 Free PMC article.
-
Structural biology of ex vivo mammalian prions.J Biol Chem. 2022 Aug;298(8):102181. doi: 10.1016/j.jbc.2022.102181. Epub 2022 Jun 23. J Biol Chem. 2022. PMID: 35752366 Free PMC article. Review.
-
Evolving views in prion glycosylation: functional and pathological implications.Biochimie. 2003 Jan-Feb;85(1-2):33-45. doi: 10.1016/s0300-9084(03)00040-3. Biochimie. 2003. PMID: 12765773 Review.
Cited by
-
Melatonin: Regulation of Prion Protein Phase Separation in Cancer Multidrug Resistance.Molecules. 2022 Jan 21;27(3):705. doi: 10.3390/molecules27030705. Molecules. 2022. PMID: 35163973 Free PMC article. Review.
-
Insight From Animals Resistant to Prion Diseases: Deciphering the Genotype - Morphotype - Phenotype Code for the Prion Protein.Front Cell Neurosci. 2020 Aug 18;14:254. doi: 10.3389/fncel.2020.00254. eCollection 2020. Front Cell Neurosci. 2020. PMID: 33013324 Free PMC article. Review.
-
Anchorless risk or released benefit? An updated view on the ADAM10-mediated shedding of the prion protein.Cell Tissue Res. 2023 Apr;392(1):215-234. doi: 10.1007/s00441-022-03582-4. Epub 2022 Jan 27. Cell Tissue Res. 2023. PMID: 35084572 Free PMC article. Review.
-
Short and sweet: How glycans impact prion conversion, cofactor interactions, and cross-species transmission.PLoS Pathog. 2021 Jan 14;17(1):e1009123. doi: 10.1371/journal.ppat.1009123. eCollection 2021 Jan. PLoS Pathog. 2021. PMID: 33444414 Free PMC article. No abstract available.
References
-
- Aguilar-Calvo P, Sevillano AM, Bapat J, Soldau K, Sandoval DR, Altmeppen HC, Linsenmeier L, Pizzo DP, Geschwind MD, Sanchez H et al. (2019) Shortening heparan sulfate chains prolongs survival and reduces parenchymal plaques in prion disease caused by mobile, ADAM10-cleaved prions. Acta Neuropathol: Doi 10.1007/s00401-019-02085-x - DOI - PMC - PubMed
-
- Baldwin MA, Stahl N, Reinders LG, Gibson BW, Prusiner SB, Burlingame AL (1990) Permethylation and tandem mass spectrometry of oligosaccharides having free hexosamine: analysis of the glycoinositol phospholipid anchor glycan from the scrapie prion protein. Anal Biochem 191: 174–182 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
