

SPG7 mutations in amyotrophic lateral sclerosis: a genetic link to hereditary spastic paraplegia
- PMID: 32447552
- PMCID: PMC7419373
- DOI: 10.1007/s00415-020-09861-w
SPG7 mutations in amyotrophic lateral sclerosis: a genetic link to hereditary spastic paraplegia
Abstract
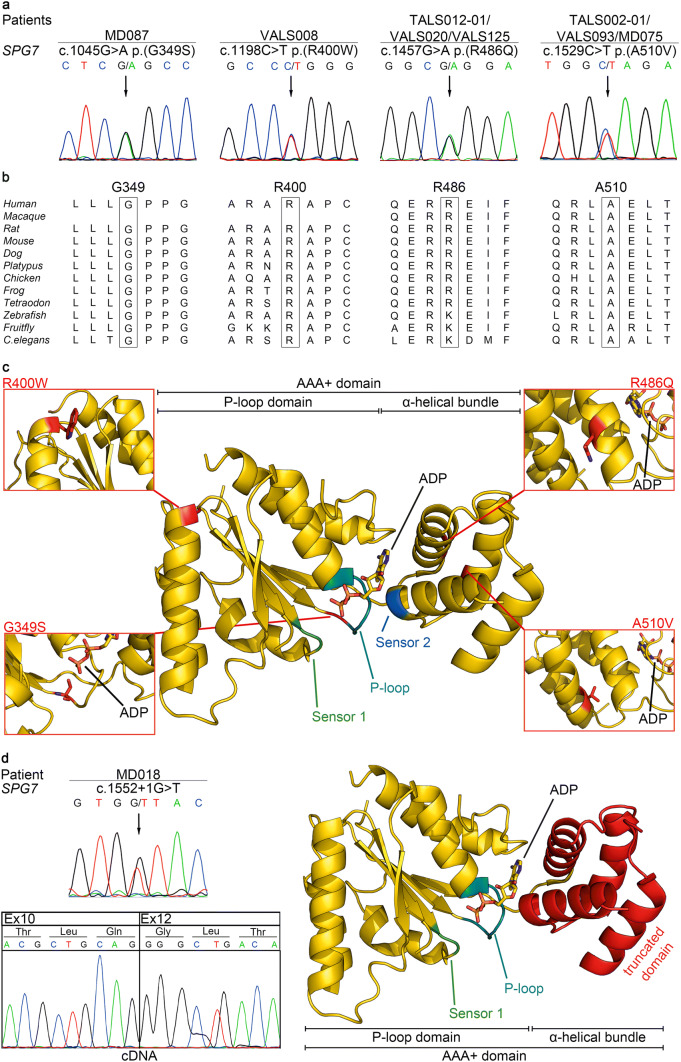
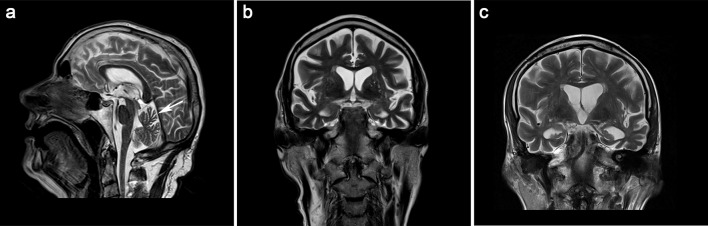
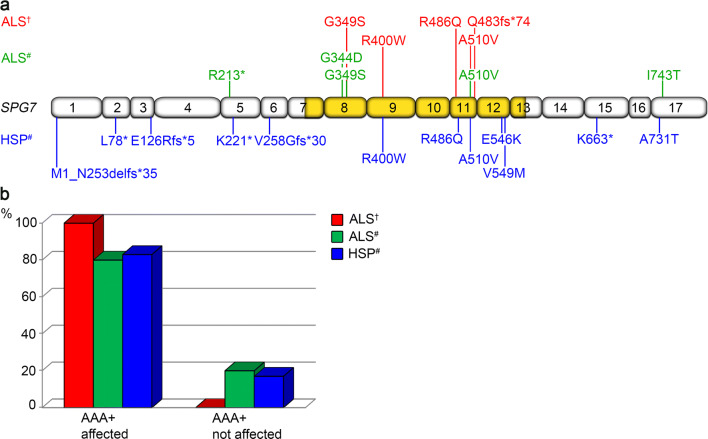
Amyotrophic lateral sclerosis (ALS) and hereditary spastic paraplegia (HSP) are motor neuron diseases sharing clinical, pathological, and genetic similarities. While biallelic SPG7 mutations are known to cause recessively inherited HSP, heterozygous SPG7 mutations have repeatedly been identified in HSP and recently also in ALS cases. However, the frequency and clinical impact of rare SPG7 variants have not been studied in a larger ALS cohort. Here, whole-exome (WES) or targeted SPG7 sequencing was done in a cohort of 214 European ALS patients. The consequences of a splice site variant were analyzed on the mRNA level. The resulting protein alterations were visualized in a crystal structure model. All patients were subjected to clinical, electrophysiological, and neuroradiological characterization. In 9 of 214 (4.2%) ALS cases, we identified five different rare heterozygous SPG7 variants, all of which were previously reported in patients with HSP or ALS. All detected SPG7 variants affect the AAA+ domain of the encoded mitochondrial metalloprotease paraplegin and impair its stability or function according to predictions from mRNA analysis or crystal structure modeling. ALS patients with SPG7 mutations more frequently presented with cerebellar symptoms, flail arm or leg syndrome compared to those without SPG7 mutations, and showed a partial clinical overlap with HSP. Brain MRI findings in SPG7 mutation carriers included cerebellar atrophy and patterns suggestive of frontotemporal dementia. Collectively, our findings suggest that SPG7 acts as a genetic risk factor for ALS. ALS patients carrying SPG7 mutations present with distinct features overlapping with HSP, particularly regarding cerebellar findings.
Keywords: Amyotrophic lateral sclerosis; Hereditary spastic paraplegia; Motor neuron disease; SPG7; Whole-exome sequencing.
Conflict of interest statement
AO received honoraria from Biogen; MPW received honoraria from Bayer, Biogen, Biologix, Celgene, Genilac, Imcyse, IXICO, Medison, Merck-Serono, Novartis, Roche, Sanofi-Genzyme, Spinger Healthcare, and Teva; PR received honoraria from Biogen and IXICO; SP received honoraria from Biogen, Cytokinetics, Inc., Desitin Pharma, Novartis, Roche, and Teva. MW, AF, JW, IL, AS, BA, AC, MP, and RGW report no conflict of interest.
Figures
Similar articles
-
Evidence for Non-Mendelian Inheritance in Spastic Paraplegia 7.Mov Disord. 2021 Jul;36(7):1664-1675. doi: 10.1002/mds.28528. Epub 2021 Feb 17. Mov Disord. 2021. PMID: 33598982
-
Identification of novel compound heterozygous SPG7 mutations-related hereditary spastic paraplegia in a Chinese family: a case report.BMC Neurol. 2018 Nov 29;18(1):196. doi: 10.1186/s12883-018-1199-9. BMC Neurol. 2018. PMID: 30497413 Free PMC article.
-
An SPG7 mutation as a novel cause of monogenic progressive muscular atrophy.Neurol Sci. 2023 Sep;44(9):3303-3305. doi: 10.1007/s10072-023-06867-w. Epub 2023 May 22. Neurol Sci. 2023. PMID: 37213040
-
Genetic architecture of motor neuron diseases.J Neurol Sci. 2022 Mar 15;434:120099. doi: 10.1016/j.jns.2021.120099. Epub 2021 Dec 22. J Neurol Sci. 2022. PMID: 34965490 Review.
-
Primary lateral sclerosis, hereditary spastic paraplegia and amyotrophic lateral sclerosis: discrete entities or spectrum?Amyotroph Lateral Scler Other Motor Neuron Disord. 2005 Mar;6(1):8-16. doi: 10.1080/14660820410021267. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005. PMID: 16036421 Review.
Cited by
-
High-Throughput Genetic Testing in ALS: The Challenging Path of Variant Classification Considering the ACMG Guidelines.Genes (Basel). 2020 Sep 24;11(10):1123. doi: 10.3390/genes11101123. Genes (Basel). 2020. PMID: 32987860 Free PMC article.
-
A SUMO4 initiator codon variant in amyotrophic lateral sclerosis reduces SUMO4 expression and alters stress granule dynamics.J Neurol. 2022 Sep;269(9):4863-4871. doi: 10.1007/s00415-022-11126-7. Epub 2022 May 3. J Neurol. 2022. PMID: 35503374 Free PMC article.
-
Mechanism and treatment of intracerebral hemorrhage focus on mitochondrial permeability transition pore.Front Mol Neurosci. 2024 Jul 31;17:1423132. doi: 10.3389/fnmol.2024.1423132. eCollection 2024. Front Mol Neurosci. 2024. PMID: 39156127 Free PMC article. Review.
-
Hereditary Spastic Paraplegia: An Update.Int J Mol Sci. 2022 Feb 1;23(3):1697. doi: 10.3390/ijms23031697. Int J Mol Sci. 2022. PMID: 35163618 Free PMC article. Review.
-
Hereditary spastic paraparesis type 18 (SPG18): new ERLIN2 variants in a series of Italian patients, shedding light upon genetic and phenotypic variability.Neurol Sci. 2024 Aug;45(8):3845-3852. doi: 10.1007/s10072-024-07423-w. Epub 2024 Mar 1. Neurol Sci. 2024. PMID: 38427163 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
