

Identification of a novel toxicophore in anti-cancer chemotherapeutics that targets mitochondrial respiratory complex I
- PMID: 32432547
- PMCID: PMC7316505
- DOI: 10.7554/eLife.55845
Identification of a novel toxicophore in anti-cancer chemotherapeutics that targets mitochondrial respiratory complex I
Abstract
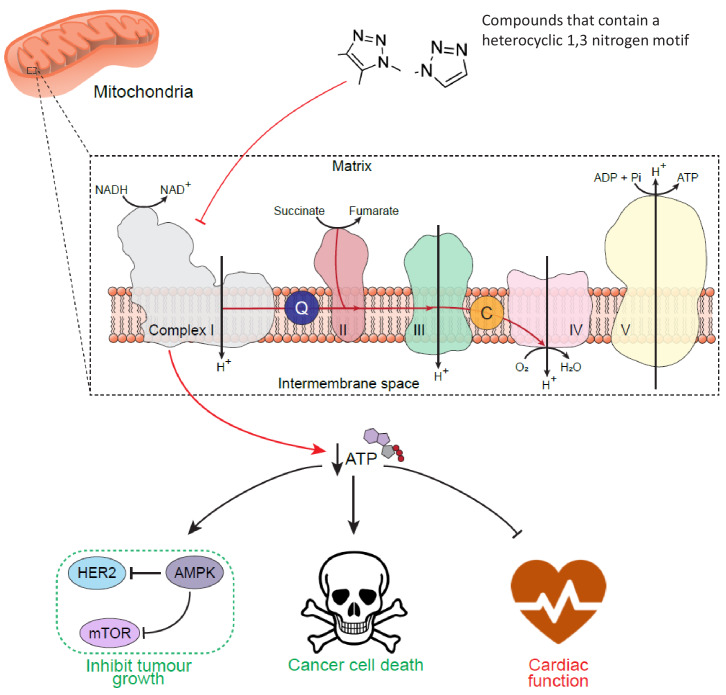
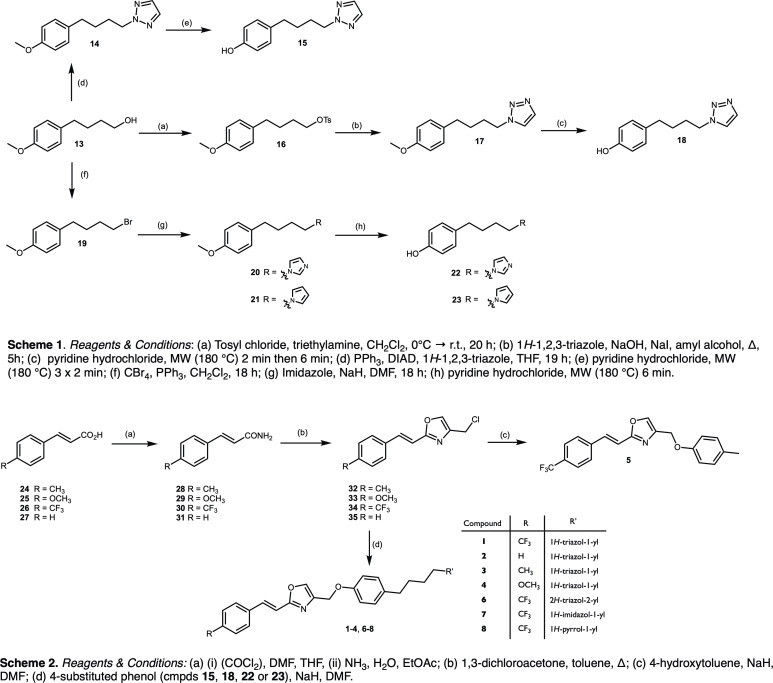
Disruption of mitochondrial function selectively targets tumour cells that are dependent on oxidative phosphorylation. However, due to their high energy demands, cardiac cells are disproportionately targeted by mitochondrial toxins resulting in a loss of cardiac function. An analysis of the effects of mubritinib on cardiac cells showed that this drug did not inhibit HER2 as reported, but directly inhibits mitochondrial respiratory complex I, reducing cardiac-cell beat rate, with prolonged exposure resulting in cell death. We used a library of chemical variants of mubritinib and showed that modifying the 1H-1,2,3-triazole altered complex I inhibition, identifying the heterocyclic 1,3-nitrogen motif as the toxicophore. The same toxicophore is present in a second anti-cancer therapeutic carboxyamidotriazole (CAI) and we demonstrate that CAI also functions through complex I inhibition, mediated by the toxicophore. Complex I inhibition is directly linked to anti-cancer cell activity, with toxicophore modification ablating the desired effects of these compounds on cancer cell proliferation and apoptosis.
Keywords: biochemistry; cancer biology; cardiac liability; chemical biology; human; mitochondria; toxicophore.
Plain language summary
The pharmaceutical industry needs to make safe and effective drugs. At the same time this industry is under pressure to keep the costs of developing these drugs at an acceptable level. Drugs work by interacting with and typically blocking a specific target, such as a protein in a particular type of cell. Sometimes, however, drugs also bind other unexpected targets. These “off-target” effects can be the reason for a drug’s toxicity, and it is important – both for the benefit of patients and the money that can be saved when developing drugs – to identify how drugs cause toxic side effects. The earlier researchers detect off-target effects, the better. Recent data has suggested that an anti-cancer drug called mubritinib has off-target effects on the compartments within cells that provide the cell with most of their energy, the mitochondria. This drug’s intended target is a protein called HER2, which is found in large amounts on the surfaces of some breast cancer cells. Yet if mubritinib has this off-target effect on mitochondria, it may be harmful to other cells including heart cells because the heart is an organ that needs a large amount of energy from its mitochondria. Stephenson et al. have now performed experiments to show that mubritinib does not actually interact with HER2 at all, but only targets mitochondria. The effect of mubritinib as an anti-cancer drug is therefore only due to its activity against mitochondria. Digging deeper into the chemistry revealed the small parts of its chemical structure that was responsible for mubritinib’s toxicity against heart cells, the so-called toxic substructure. Another anti-cancer drug called carboxyamidotriazole also has the same toxic substructure. Carboxyamidotriazole is supposed to stop cells from taking up calcium ions, but a final set of experiments demonstrated that this drug also only acts by inhibiting mitochondria. Often there is not enough information about many drugs’ substructures, meaning off-target effects and toxicities cannot be predicted. The pharmaceutical industry will now be able to benefit from this new knowledge about the toxic substructures within some drugs. This research may also help patients who take mubritinib or carboxyamidotriazole, because their doctors will have to check for side effects on the heart more carefully.
© 2020, Stephenson et al.
Conflict of interest statement
ZS, RH, KP, SM, RH, RS, IC, TA, MS, MM, PF, JH, BK, AW No competing interests declared
Figures
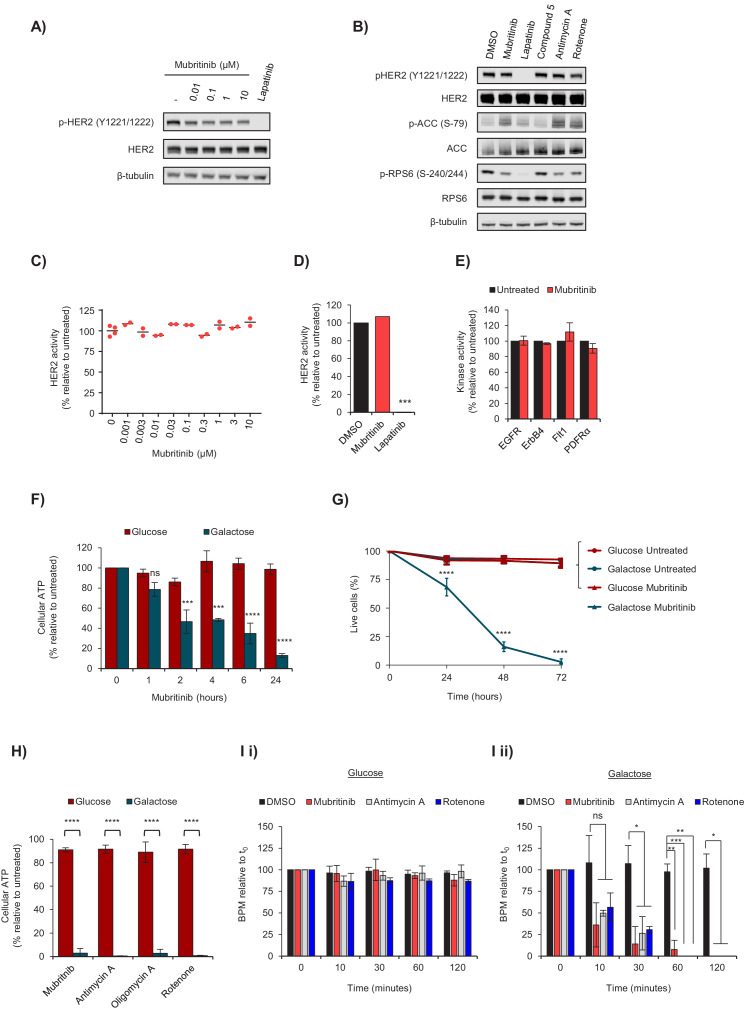

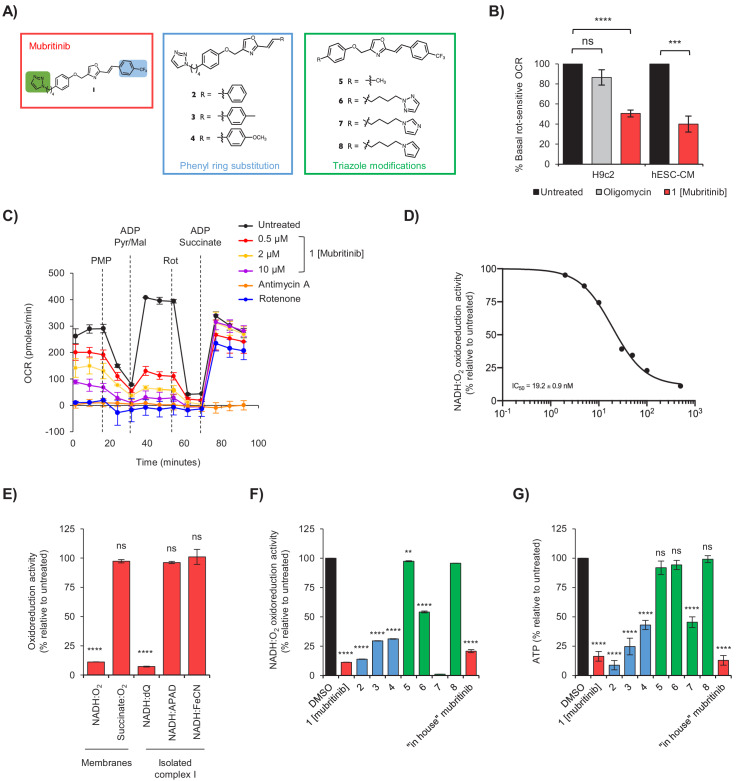
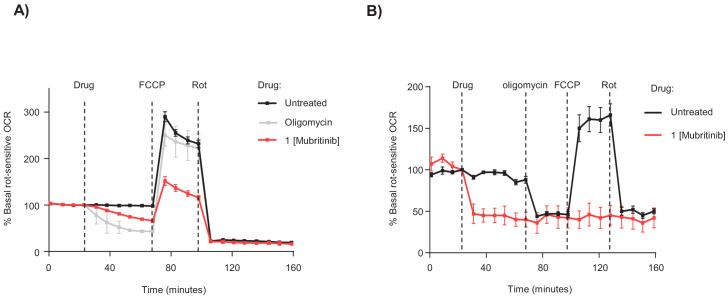
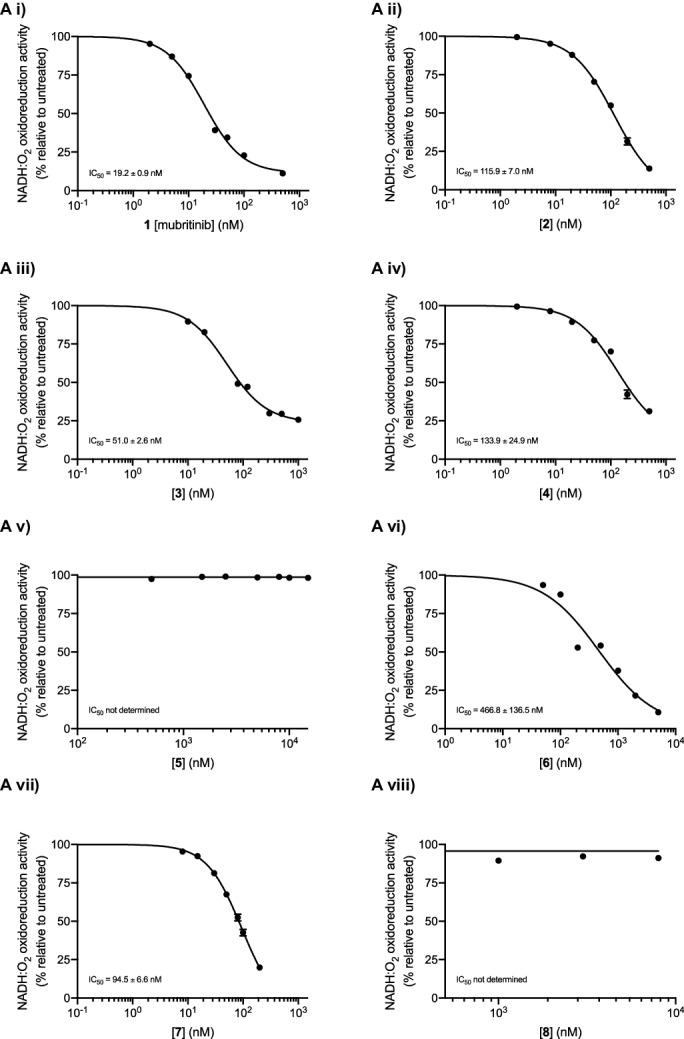
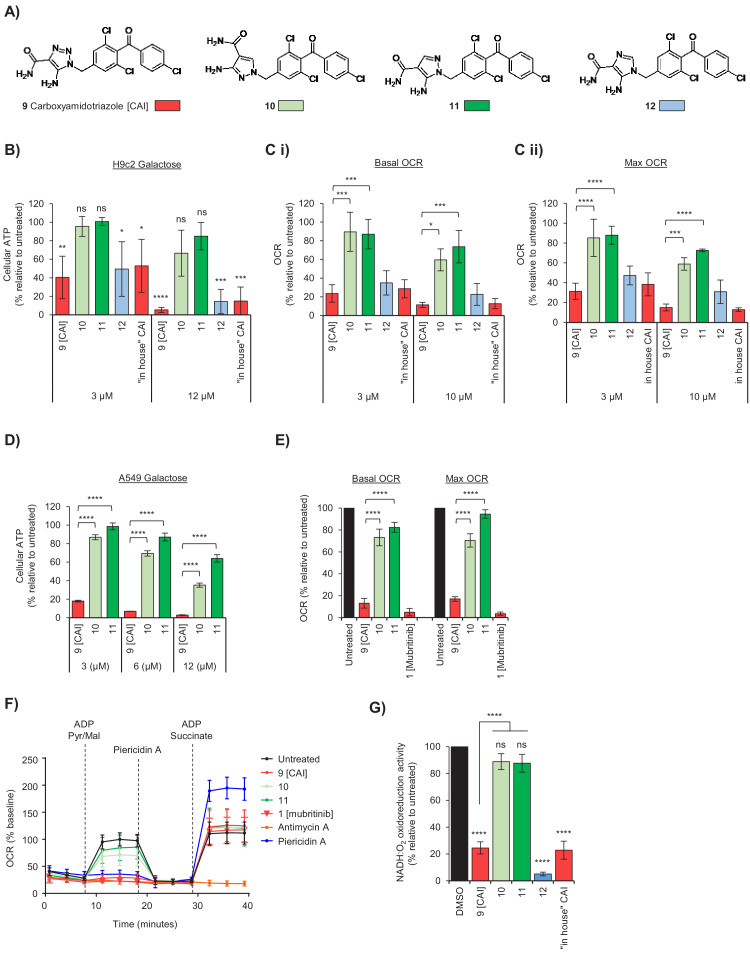


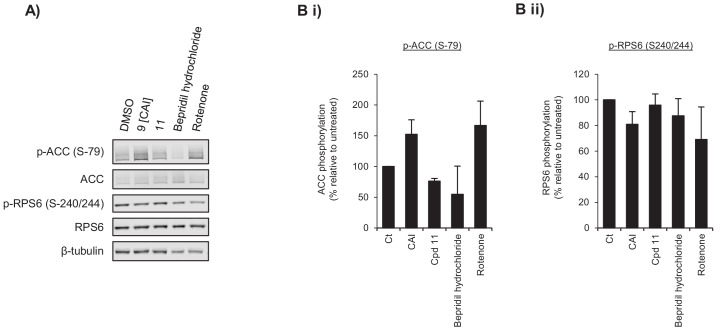
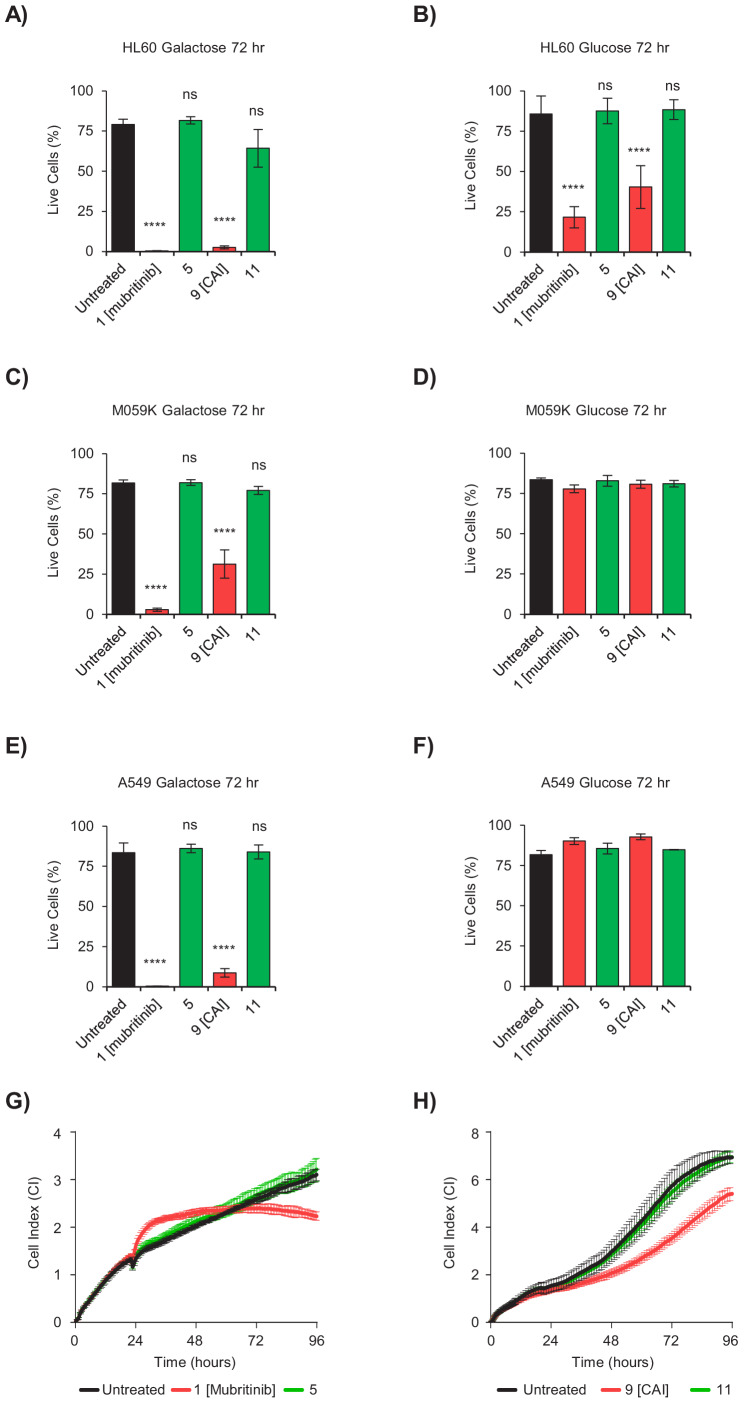
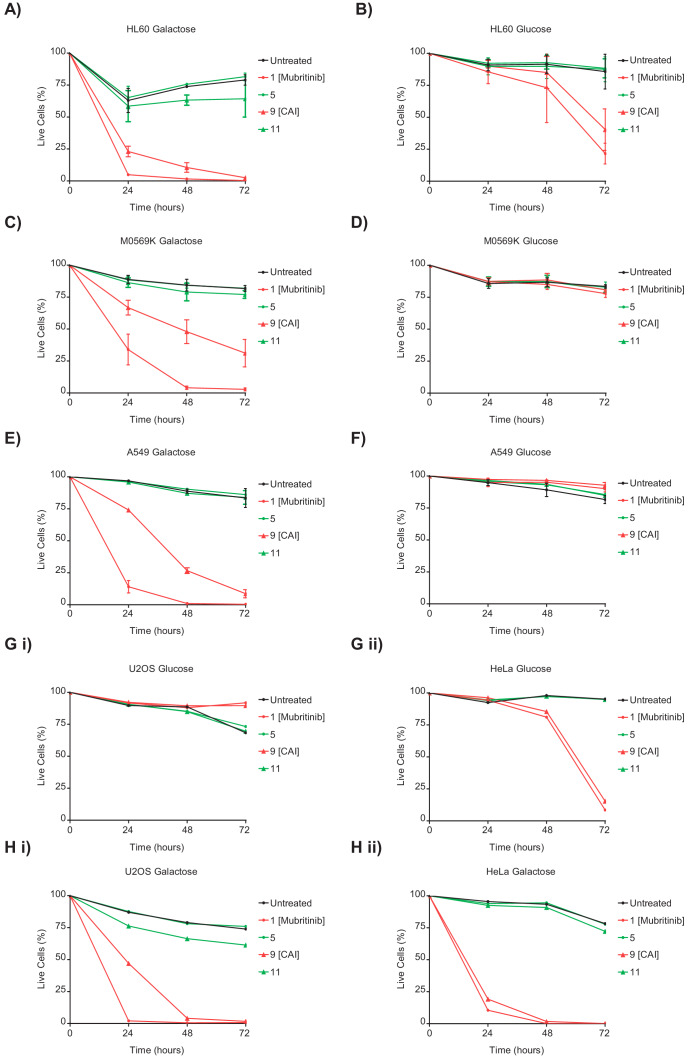

Comment in
-
The mitochondrial paradox.Elife. 2020 Jun 25;9:e59140. doi: 10.7554/eLife.59140. Elife. 2020. PMID: 32583799 Free PMC article.
Similar articles
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
-
Qualitative evidence synthesis informing our understanding of people's perceptions and experiences of targeted digital communication.Cochrane Database Syst Rev. 2019 Oct 23;10(10):ED000141. doi: 10.1002/14651858.ED000141. Cochrane Database Syst Rev. 2019. PMID: 31643081 Free PMC article.
-
"I've Spent My Whole Life Striving to Be Normal": Internalized Stigma and Perceived Impact of Diagnosis in Autistic Adults.Autism Adulthood. 2023 Dec 1;5(4):423-436. doi: 10.1089/aut.2022.0066. Epub 2023 Dec 12. Autism Adulthood. 2023. PMID: 38116050 Free PMC article.
-
Using Experience Sampling Methodology to Capture Disclosure Opportunities for Autistic Adults.Autism Adulthood. 2023 Dec 1;5(4):389-400. doi: 10.1089/aut.2022.0090. Epub 2023 Dec 12. Autism Adulthood. 2023. PMID: 38116059 Free PMC article.
-
Trends in Surgical and Nonsurgical Aesthetic Procedures: A 14-Year Analysis of the International Society of Aesthetic Plastic Surgery-ISAPS.Aesthetic Plast Surg. 2024 Oct;48(20):4217-4227. doi: 10.1007/s00266-024-04260-2. Epub 2024 Aug 5. Aesthetic Plast Surg. 2024. PMID: 39103642 Review.
Cited by
-
Mubritinib enhanced the inhibiting function of cisplatin in lung cancer by interfering with mitochondrial function.Thorac Cancer. 2022 May;13(10):1513-1524. doi: 10.1111/1759-7714.14425. Epub 2022 Apr 16. Thorac Cancer. 2022. PMID: 35429141 Free PMC article.
-
Cork-in-bottle mechanism of inhibitor binding to mammalian complex I.Sci Adv. 2021 May 14;7(20):eabg4000. doi: 10.1126/sciadv.abg4000. Print 2021 May. Sci Adv. 2021. PMID: 33990335 Free PMC article.
-
Assessing Drug-Induced Mitochondrial Toxicity in Cardiomyocytes: Implications for Preclinical Cardiac Safety Evaluation.Pharmaceutics. 2022 Jun 21;14(7):1313. doi: 10.3390/pharmaceutics14071313. Pharmaceutics. 2022. PMID: 35890211 Free PMC article. Review.
-
Binding of Natural Inhibitors to Respiratory Complex I.Pharmaceuticals (Basel). 2022 Aug 31;15(9):1088. doi: 10.3390/ph15091088. Pharmaceuticals (Basel). 2022. PMID: 36145309 Free PMC article. Review.
-
Click Chemistry in Natural Product Modification.Front Chem. 2021 Nov 17;9:774977. doi: 10.3389/fchem.2021.774977. eCollection 2021. Front Chem. 2021. PMID: 34869223 Free PMC article. Review.
References
-
- Baccelli I, Gareau Y, Lehnertz B, Gingras S, Spinella JF, Corneau S, Mayotte N, Girard S, Frechette M, Blouin-Chagnon V, Leveillé K, Boivin I, MacRae T, Krosl J, Thiollier C, Lavallée VP, Kanshin E, Bertomeu T, Coulombe-Huntington J, St-Denis C, Bordeleau ME, Boucher G, Roux PP, Lemieux S, Tyers M, Thibault P, Hébert J, Marinier A, Sauvageau G. Mubritinib targets the electron transport chain complex I and reveals the landscape of OXPHOS dependency in acute myeloid leukemia. Cancer Cell. 2019;36:84–99. doi: 10.1016/j.ccell.2019.06.003. - DOI - PubMed
-
- Bauer KS, Cude KJ, Dixon SC, Kruger EA, Figg WD. Carboxyamido-triazole inhibits angiogenesis by blocking the calcium-mediated nitric-oxide synthase-vascular endothelial growth factor pathway. The Journal of Pharmacology and Experimental Therapeutics. 2000;292:31–37. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
