

The Genomic Landscape of Intrinsic and Acquired Resistance to Cyclin-Dependent Kinase 4/6 Inhibitors in Patients with Hormone Receptor-Positive Metastatic Breast Cancer
- PMID: 32404308
- PMCID: PMC8815415
- DOI: 10.1158/2159-8290.CD-19-1390
The Genomic Landscape of Intrinsic and Acquired Resistance to Cyclin-Dependent Kinase 4/6 Inhibitors in Patients with Hormone Receptor-Positive Metastatic Breast Cancer
Abstract
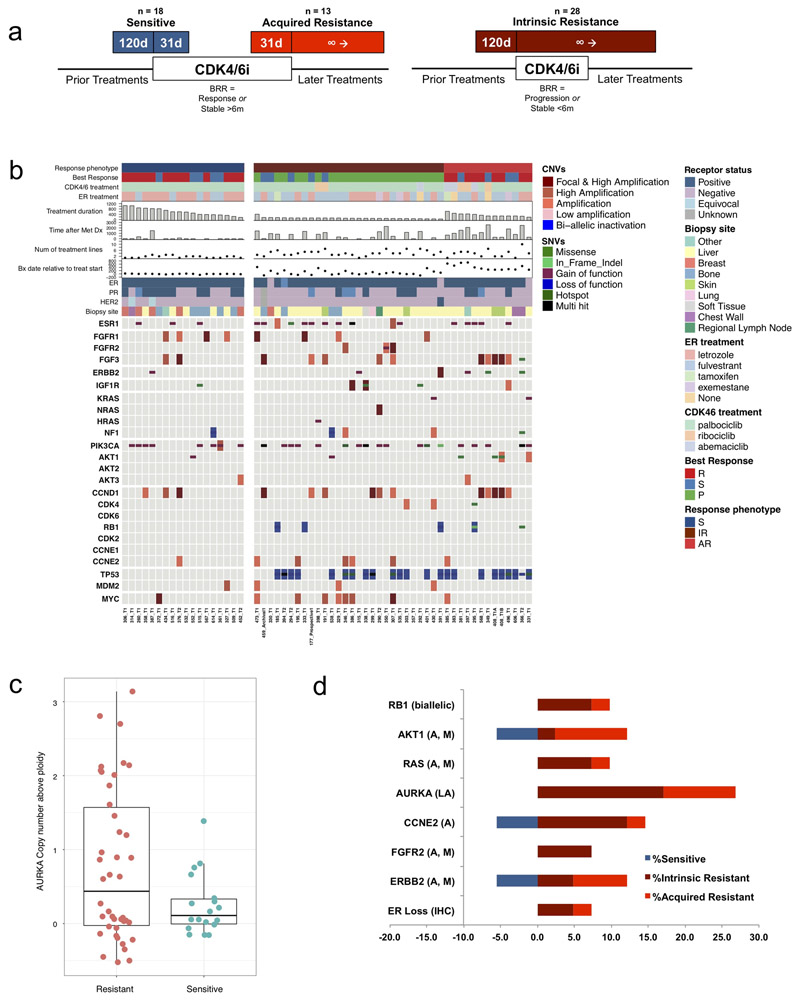
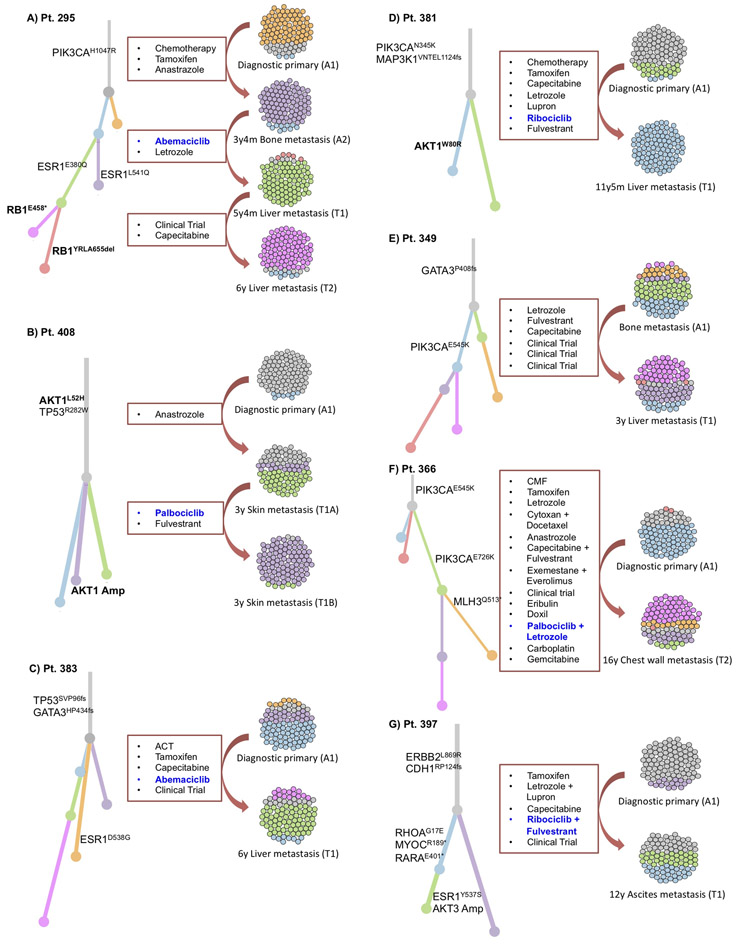
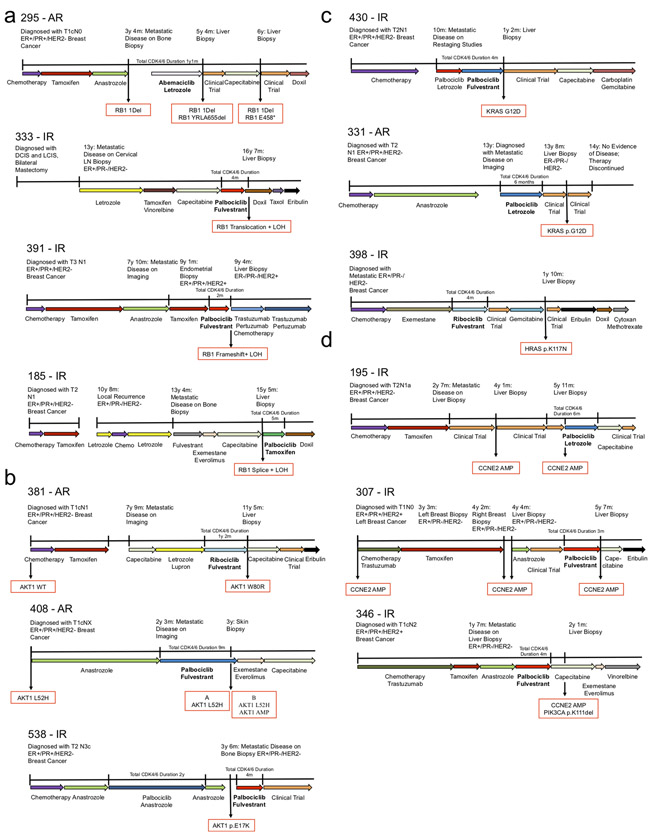
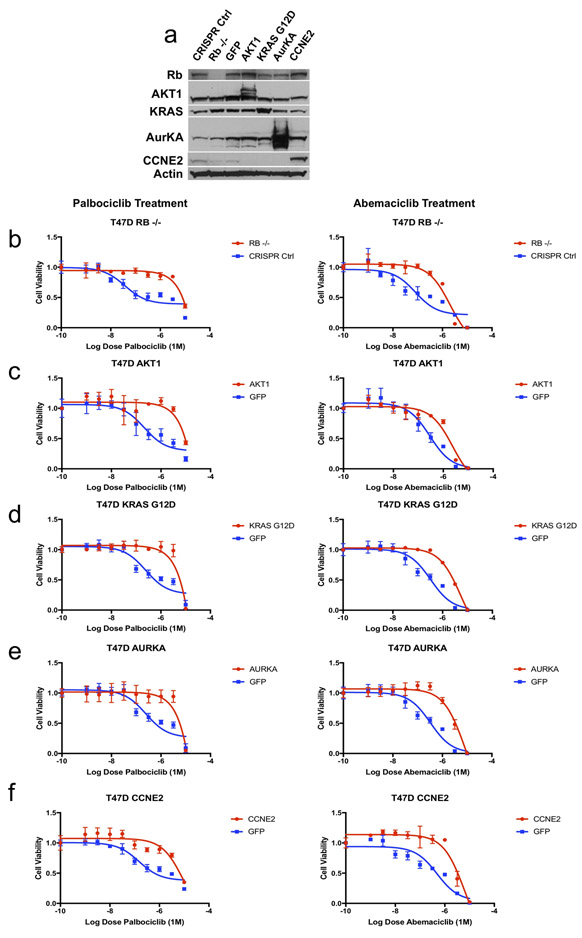
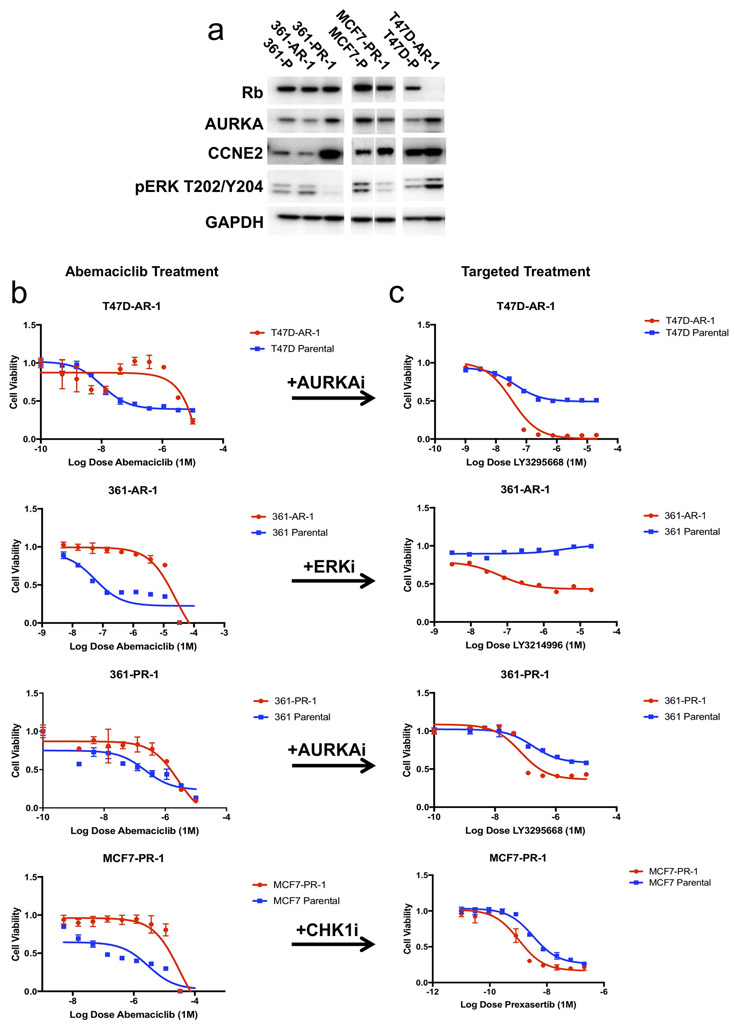
Mechanisms driving resistance to cyclin-dependent kinase 4/6 inhibitors (CDK4/6i) in hormone receptor-positive (HR+) breast cancer have not been clearly defined. Whole-exome sequencing of 59 tumors with CDK4/6i exposure revealed multiple candidate resistance mechanisms including RB1 loss, activating alterations in AKT1, RAS, AURKA, CCNE2, ERBB2, and FGFR2, and loss of estrogen receptor expression. In vitro experiments confirmed that these alterations conferred CDK4/6i resistance. Cancer cells cultured to resistance with CDK4/6i also acquired RB1, KRAS, AURKA, or CCNE2 alterations, which conferred sensitivity to AURKA, ERK, or CHEK1 inhibition. Three of these activating alterations-in AKT1, RAS, and AURKA-have not, to our knowledge, been previously demonstrated as mechanisms of resistance to CDK4/6i in breast cancer preclinically or in patient samples. Together, these eight mechanisms were present in 66% of resistant tumors profiled and may define therapeutic opportunities in patients. SIGNIFICANCE: We identified eight distinct mechanisms of resistance to CDK4/6i present in 66% of resistant tumors profiled. Most of these have a therapeutic strategy to overcome or prevent resistance in these tumors. Taken together, these findings have critical implications related to the potential utility of precision-based approaches to overcome resistance in many patients with HR+ metastatic breast cancer.This article is highlighted in the In This Issue feature, p. 1079.
©2020 American Association for Cancer Research.
Figures

Similar articles
-
Elacestrant (RAD1901) exhibits anti-tumor activity in multiple ER+ breast cancer models resistant to CDK4/6 inhibitors.Breast Cancer Res. 2019 Dec 18;21(1):146. doi: 10.1186/s13058-019-1230-0. Breast Cancer Res. 2019. PMID: 31852484 Free PMC article.
-
Practical Treatment Strategies and Future Directions After Progression While Receiving CDK4/6 Inhibition and Endocrine Therapy in Advanced HR+/HER2- Breast Cancer.Clin Breast Cancer. 2020 Feb;20(1):1-11. doi: 10.1016/j.clbc.2019.06.017. Epub 2019 Aug 23. Clin Breast Cancer. 2020. PMID: 31780379
-
Resistance to cyclin-dependent kinase (CDK) 4/6 inhibitors confers cross-resistance to other CDK inhibitors but not to chemotherapeutic agents in breast cancer cells.Breast Cancer. 2021 Jan;28(1):206-215. doi: 10.1007/s12282-020-01150-8. Epub 2020 Aug 28. Breast Cancer. 2021. PMID: 32860163 Free PMC article.
-
Therapy after cyclin-dependent kinase inhibition in metastatic hormone receptor-positive breast cancer: Resistance mechanisms and novel treatment strategies.Cancer. 2020 Aug 1;126(15):3400-3416. doi: 10.1002/cncr.32931. Epub 2020 May 19. Cancer. 2020. PMID: 32426848 Review.
-
Sequencing Endocrine Therapy for Metastatic Breast Cancer: What Do We Do After Disease Progression on a CDK4/6 Inhibitor?Curr Oncol Rep. 2020 May 16;22(6):57. doi: 10.1007/s11912-020-00917-8. Curr Oncol Rep. 2020. PMID: 32415339 Review.
Cited by
-
Safety and pharmacokinetics of vepdegestrant in Japanese patients with ER+ advanced breast cancer: a phase 1 study.Int J Clin Oncol. 2024 Nov 20. doi: 10.1007/s10147-024-02648-3. Online ahead of print. Int J Clin Oncol. 2024. PMID: 39565495
-
Mechanisms of endocrine resistance in hormone receptor-positive breast cancer.Front Oncol. 2024 Oct 31;14:1448687. doi: 10.3389/fonc.2024.1448687. eCollection 2024. Front Oncol. 2024. PMID: 39544302 Free PMC article. Review.
-
Adjuvant CDK4/6 inhibitors in hormone receptor-positive early breast cancer: one fits all?Transl Breast Cancer Res. 2024 Oct 25;5:34. doi: 10.21037/tbcr-24-41. eCollection 2024. Transl Breast Cancer Res. 2024. PMID: 39534583 Free PMC article. No abstract available.
-
A multi-modal single-cell and spatial expression map of metastatic breast cancer biopsies across clinicopathological features.Nat Med. 2024 Nov;30(11):3236-3249. doi: 10.1038/s41591-024-03215-z. Epub 2024 Oct 30. Nat Med. 2024. PMID: 39478111 Free PMC article.
-
CDK4/6 inhibition initiates cell cycle arrest by nuclear translocation of RB and induces a multistep molecular response.Cell Death Discov. 2024 Oct 26;10(1):453. doi: 10.1038/s41420-024-02218-6. Cell Death Discov. 2024. PMID: 39461947 Free PMC article.
References
-
- Finn RS, et al. Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med 375, 1925–1936 (2016). - PubMed
-
- Cristofanilli M, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol 17, 425–439 (2016). - PubMed
-
- Hortobagyi GN, et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N Engl J Med 375, 1738–1748 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
