

Killer cell proteases can target viral immediate-early proteins to control human cytomegalovirus infection in a noncytotoxic manner
- PMID: 32282833
- PMCID: PMC7179929
- DOI: 10.1371/journal.ppat.1008426
Killer cell proteases can target viral immediate-early proteins to control human cytomegalovirus infection in a noncytotoxic manner
Abstract
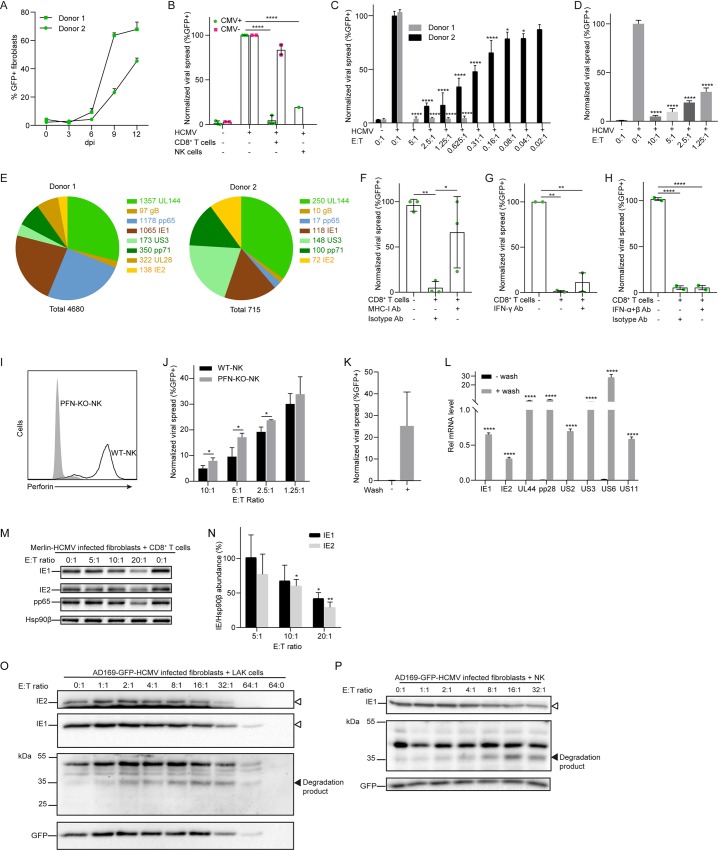
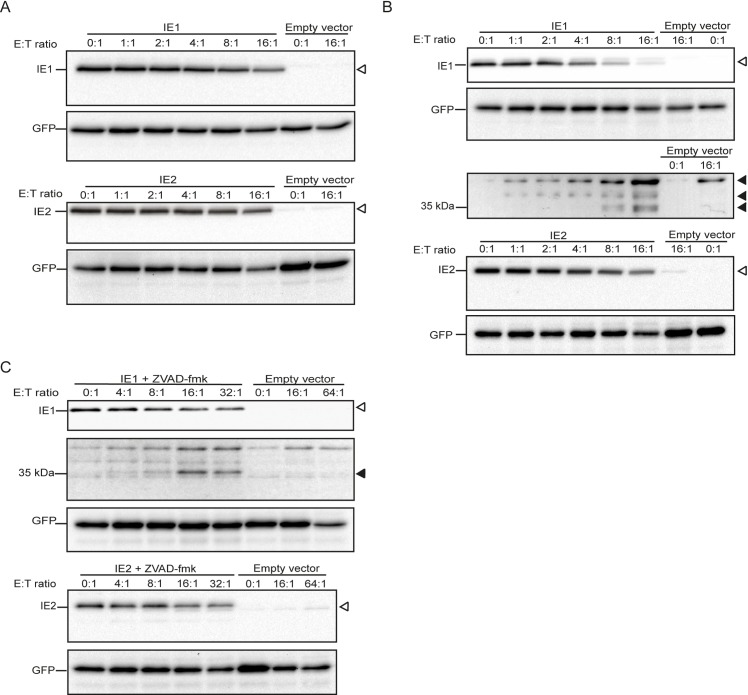
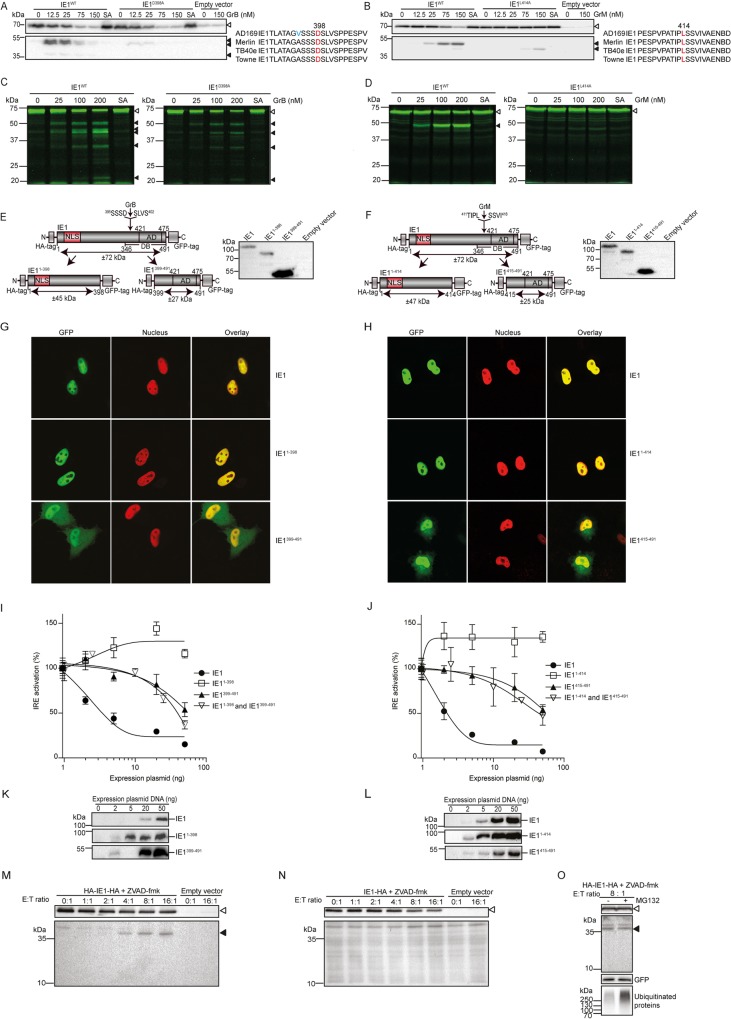
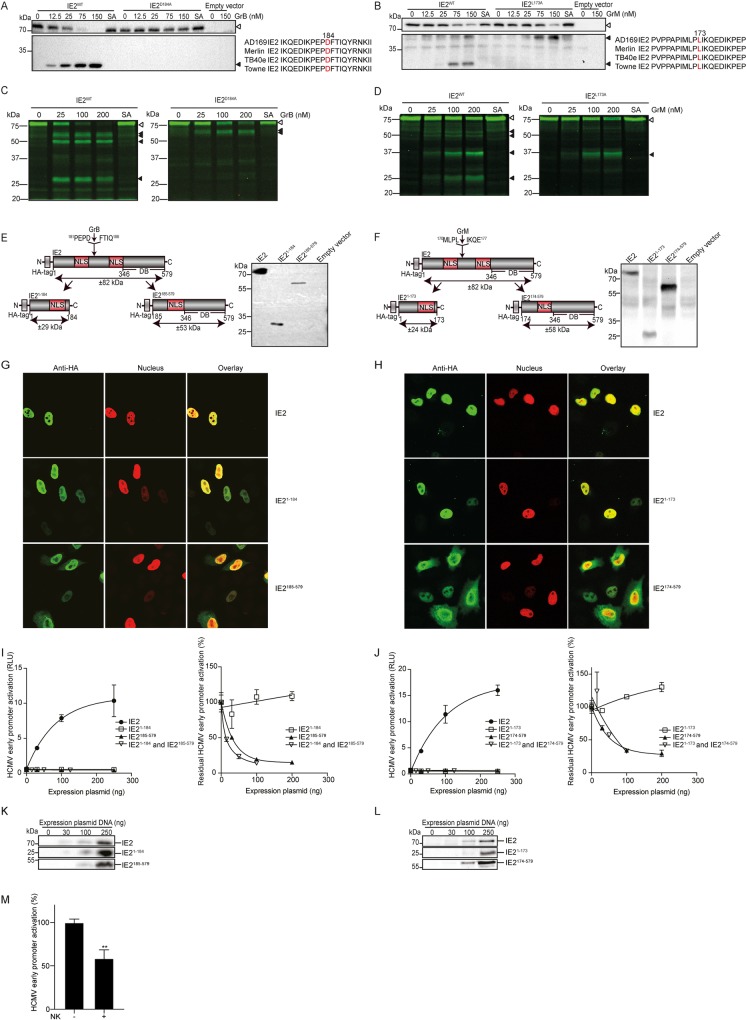
Human cytomegalovirus (HCMV) is the most frequent viral cause of congenital defects and can trigger devastating disease in immune-suppressed patients. Cytotoxic lymphocytes (CD8+ T cells and NK cells) control HCMV infection by releasing interferon-γ and five granzymes (GrA, GrB, GrH, GrK, GrM), which are believed to kill infected host cells through cleavage of intracellular death substrates. However, it has recently been demonstrated that the in vivo killing capacity of cytotoxic T cells is limited and multiple T cell hits are required to kill a single virus-infected cell. This raises the question whether cytotoxic lymphocytes can use granzymes to control HCMV infection in a noncytotoxic manner. Here, we demonstrate that (primary) cytotoxic lymphocytes can block HCMV dissemination independent of host cell death, and interferon-α/β/γ. Prior to killing, cytotoxic lymphocytes induce the degradation of viral immediate-early (IE) proteins IE1 and IE2 in HCMV-infected cells. Intriguingly, both IE1 and/or IE2 are directly proteolyzed by all human granzymes, with GrB and GrM being most efficient. GrB and GrM cleave IE1 after Asp398 and Leu414, respectively, likely resulting in IE1 aberrant cellular localization, IE1 instability, and functional impairment of IE1 to interfere with the JAK-STAT signaling pathway. Furthermore, GrB and GrM cleave IE2 after Asp184 and Leu173, respectively, resulting in IE2 aberrant cellular localization and functional abolishment of IE2 to transactivate the HCMV UL112 early promoter. Taken together, our data indicate that cytotoxic lymphocytes can also employ noncytotoxic ways to control HCMV infection, which may be explained by granzyme-mediated targeting of indispensable viral proteins during lytic infection.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
Granzyme M targets host cell hnRNP K that is essential for human cytomegalovirus replication.Cell Death Differ. 2013 Mar;20(3):419-29. doi: 10.1038/cdd.2012.132. Epub 2012 Oct 26. Cell Death Differ. 2013. PMID: 23099853 Free PMC article.
-
Noncytotoxic inhibition of cytomegalovirus replication through NK cell protease granzyme M-mediated cleavage of viral phosphoprotein 71.J Immunol. 2010 Dec 15;185(12):7605-13. doi: 10.4049/jimmunol.1001503. Epub 2010 Nov 8. J Immunol. 2010. PMID: 21059895
-
Disruption of PML-associated nuclear bodies by IE1 correlates with efficient early stages of viral gene expression and DNA replication in human cytomegalovirus infection.Virology. 2000 Aug 15;274(1):39-55. doi: 10.1006/viro.2000.0448. Virology. 2000. PMID: 10936087
-
Bright and Early: Inhibiting Human Cytomegalovirus by Targeting Major Immediate-Early Gene Expression or Protein Function.Viruses. 2020 Jan 16;12(1):110. doi: 10.3390/v12010110. Viruses. 2020. PMID: 31963209 Free PMC article. Review.
-
Cell death-independent functions of granzymes: hit viruses where it hurts.Rev Med Virol. 2011 Sep;21(5):301-14. doi: 10.1002/rmv.697. Epub 2011 Jun 29. Rev Med Virol. 2011. PMID: 21714121 Review.
Cited by
-
Oxidative and Non-Oxidative Antimicrobial Activities of the Granzymes.Front Immunol. 2021 Oct 11;12:750512. doi: 10.3389/fimmu.2021.750512. eCollection 2021. Front Immunol. 2021. PMID: 34707614 Free PMC article. Review.
-
microRNA, a Subtle Indicator of Human Cytomegalovirus against Host Immune Cells.Vaccines (Basel). 2022 Jan 19;10(2):144. doi: 10.3390/vaccines10020144. Vaccines (Basel). 2022. PMID: 35214602 Free PMC article. Review.
-
Assessing Anti-HCMV Cell Mediated Immune Responses in Transplant Recipients and Healthy Controls Using a Novel Functional Assay.Front Cell Infect Microbiol. 2020 Jun 26;10:275. doi: 10.3389/fcimb.2020.00275. eCollection 2020. Front Cell Infect Microbiol. 2020. PMID: 32670891 Free PMC article.
-
Noncytotoxic functions of killer cell granzymes in viral infections.PLoS Pathog. 2021 Sep 16;17(9):e1009818. doi: 10.1371/journal.ppat.1009818. eCollection 2021 Sep. PLoS Pathog. 2021. PMID: 34529743 Free PMC article. Review.
-
Monoclonal antibodies targeting nonstructural viral antigens can activate ADCC against human cytomegalovirus.J Clin Invest. 2021 Feb 15;131(4):e139296. doi: 10.1172/JCI139296. J Clin Invest. 2021. PMID: 33586678 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
