

Activation of JNK signaling promotes all- trans- retinal-induced photoreceptor apoptosis in mice
- PMID: 32265302
- PMCID: PMC7242700
- DOI: 10.1074/jbc.RA120.013189
Activation of JNK signaling promotes all- trans- retinal-induced photoreceptor apoptosis in mice
Abstract
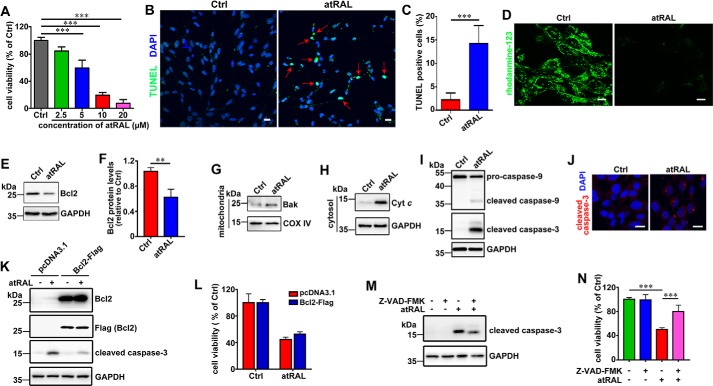
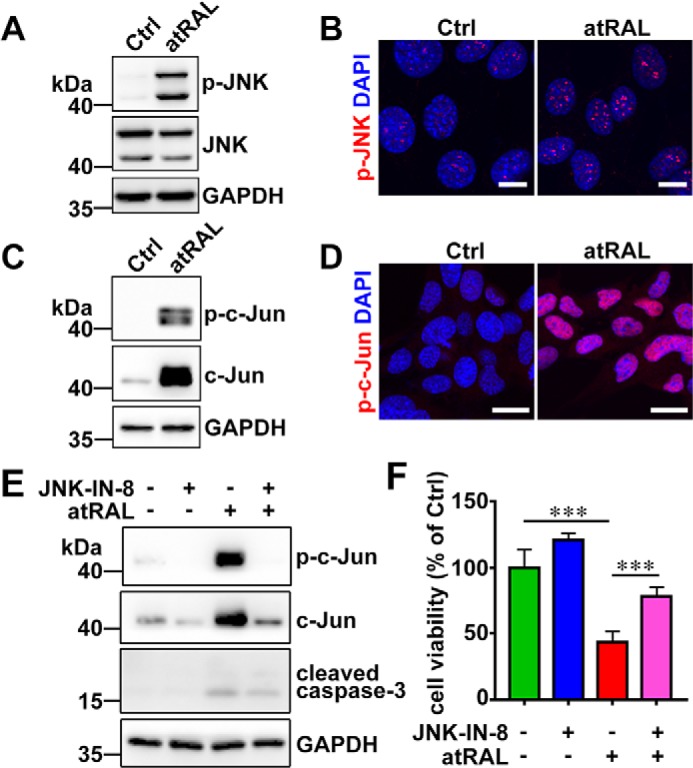
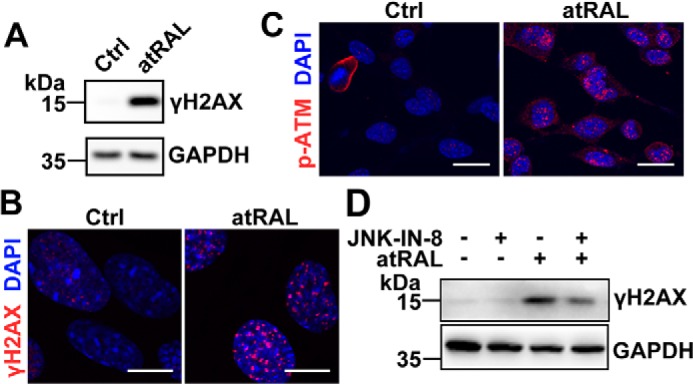
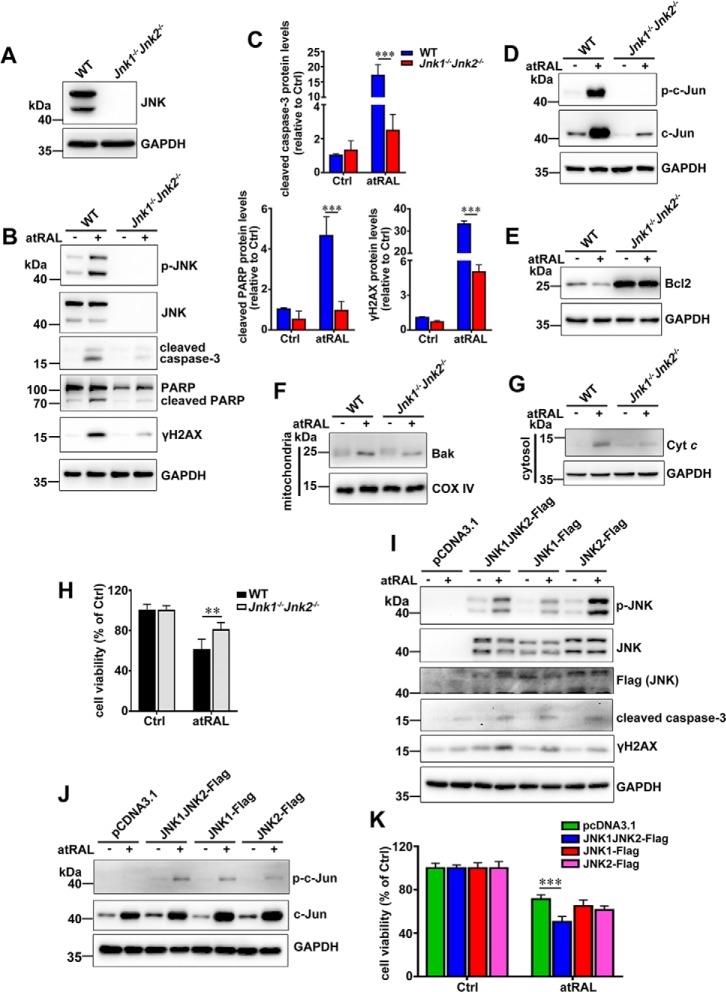
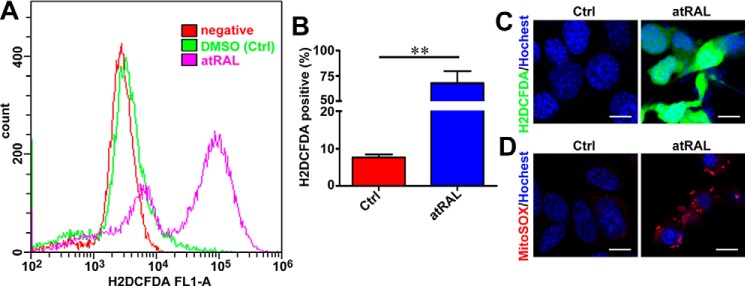
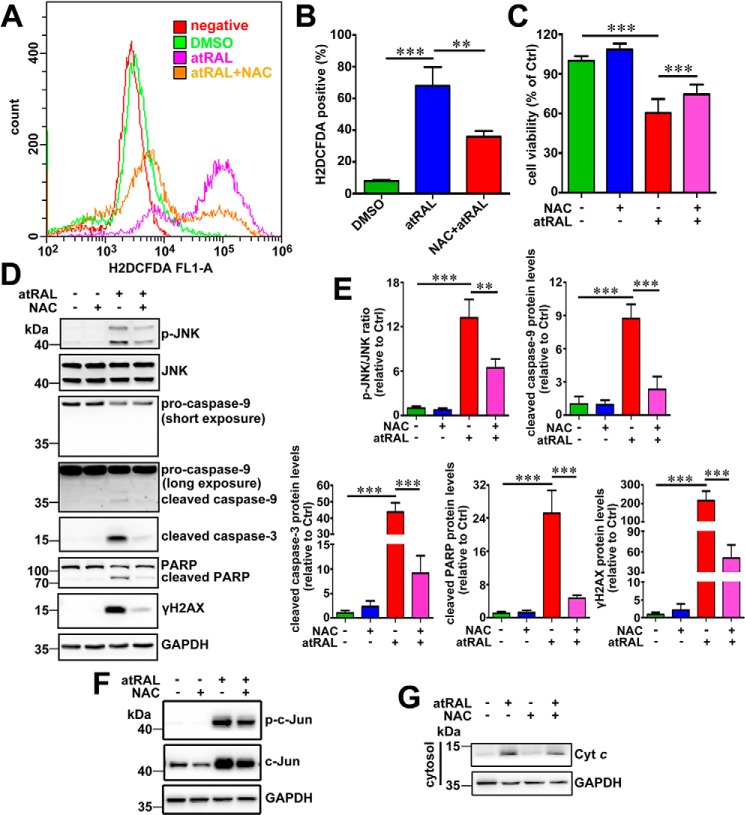
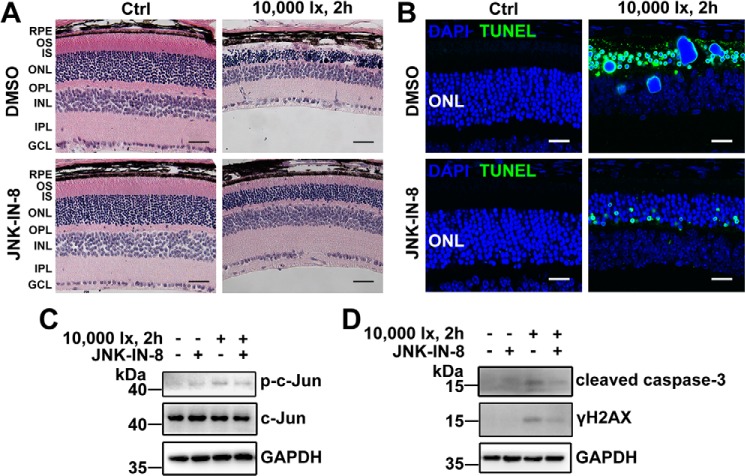
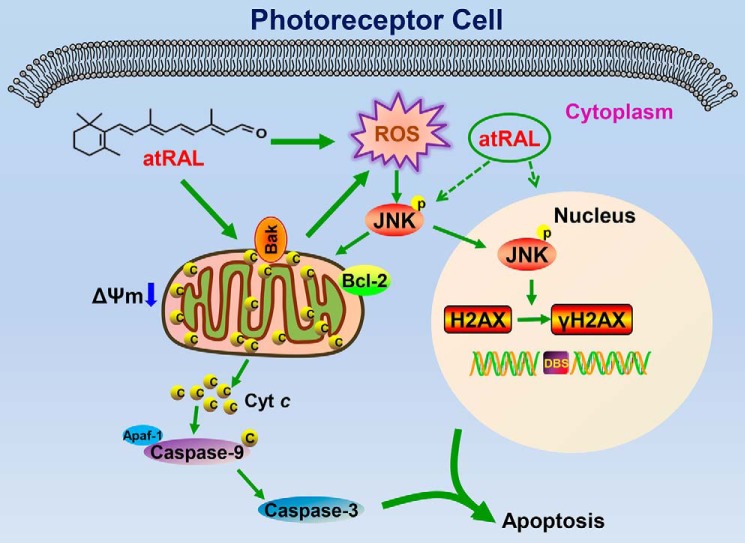
Disrupted clearance of all-trans-retinal (atRAL), a component of the visual (retinoid) cycle in the retina, may cause photoreceptor atrophy in autosomal recessive Stargardt disease (STGD1) and dry age-related macular degeneration (AMD). However, the mechanisms underlying atRAL-induced photoreceptor loss remain elusive. Here, we report that atRAL activates c-Jun N-terminal kinase (JNK) signaling at least partially through reactive oxygen species production, which promoted mitochondria-mediated caspase- and DNA damage-dependent apoptosis in photoreceptor cells. Damage to mitochondria in atRAL-exposed photoreceptor cells resulted from JNK activation, leading to decreased expression of Bcl2 apoptosis regulator (Bcl2), increased Bcl2 antagonist/killer (Bak) levels, and cytochrome c (Cyt c) release into the cytosol. Cytosolic Cyt c specifically provoked caspase-9 and caspase-3 activation and thereby initiated apoptosis. Phosphorylation of JNK in atRAL-loaded photoreceptor cells induced the appearance of γH2AX, a sensitive marker for DNA damage, and was also associated with apoptosis onset. Suppression of JNK signaling protected photoreceptor cells against atRAL-induced apoptosis. Moreover, photoreceptor cells lacking Jnk1 and Jnk2 genes were more resistant to atRAL-associated cytotoxicity. The Abca4-/-Rdh8-/- mouse model displays defects in atRAL clearance that are characteristic of STGD1 and dry AMD. We found that JNK signaling was activated in the neural retina of light-exposed Abca4-/-Rdh8-/- mice. Of note, intraperitoneal administration of JNK-IN-8, which inhibits JNK signaling, effectively ameliorated photoreceptor degeneration and apoptosis in light-exposed Abca4-/-Rdh8-/- mice. We propose that pharmacological inhibition of JNK signaling may represent a therapeutic strategy for preventing photoreceptor loss in retinopathies arising from atRAL overload.
Keywords: DNA damage; apoptosis; photoreceptor; retina; retinal metabolism.
© 2020 Liao et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures

Similar articles
-
Ferroptosis drives photoreceptor degeneration in mice with defects in all-trans-retinal clearance.J Biol Chem. 2021 Jan-Jun;296:100187. doi: 10.1074/jbc.RA120.015779. Epub 2020 Dec 20. J Biol Chem. 2021. PMID: 33334878 Free PMC article.
-
eIF2α incites photoreceptor cell and retina damage by all-trans-retinal.J Biol Chem. 2023 May;299(5):104686. doi: 10.1016/j.jbc.2023.104686. Epub 2023 Apr 7. J Biol Chem. 2023. PMID: 37031820 Free PMC article.
-
Gasdermin E mediates photoreceptor damage by all-trans-retinal in the mouse retina.J Biol Chem. 2022 Feb;298(2):101553. doi: 10.1016/j.jbc.2021.101553. Epub 2021 Dec 29. J Biol Chem. 2022. PMID: 34973334 Free PMC article.
-
Structure and function of ABCA4 and its role in the visual cycle and Stargardt macular degeneration.Prog Retin Eye Res. 2022 Jul;89:101036. doi: 10.1016/j.preteyeres.2021.101036. Epub 2021 Dec 23. Prog Retin Eye Res. 2022. PMID: 34954332 Review.
-
The Roles of c-Jun N-Terminal Kinase (JNK) in Infectious Diseases.Int J Mol Sci. 2021 Sep 6;22(17):9640. doi: 10.3390/ijms22179640. Int J Mol Sci. 2021. PMID: 34502556 Free PMC article. Review.
Cited by
-
Ferroptosis drives photoreceptor degeneration in mice with defects in all-trans-retinal clearance.J Biol Chem. 2021 Jan-Jun;296:100187. doi: 10.1074/jbc.RA120.015779. Epub 2020 Dec 20. J Biol Chem. 2021. PMID: 33334878 Free PMC article.
-
Eye Drop with Fas-Blocking Peptide Attenuates Age-Related Macular Degeneration.Cells. 2024 Mar 20;13(6):548. doi: 10.3390/cells13060548. Cells. 2024. PMID: 38534392 Free PMC article.
-
Effect of palmitoylethanolamide on degeneration of a human-derived retinal pigment epithelial cell induced by all-trans retinal.Int J Ophthalmol. 2023 Feb 18;16(2):191-200. doi: 10.18240/ijo.2023.02.04. eCollection 2023. Int J Ophthalmol. 2023. PMID: 36816211 Free PMC article.
-
Crocin Protects the 661W Murine Photoreceptor Cell Line against the Toxic Effects of All-Trans-Retinal.Int J Mol Sci. 2024 Sep 20;25(18):10124. doi: 10.3390/ijms251810124. Int J Mol Sci. 2024. PMID: 39337609 Free PMC article.
-
Transcriptome Sequencing Reveals Tgf-β-Mediated Noncoding RNA Regulatory Mechanisms Involved in DNA Damage in the 661W Photoreceptor Cell Line.Genes (Basel). 2022 Nov 17;13(11):2140. doi: 10.3390/genes13112140. Genes (Basel). 2022. PMID: 36421815 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
