

Down-regulation of AMPK signaling pathway rescues hearing loss in TFB1 transgenic mice and delays age-related hearing loss
- PMID: 32240104
- PMCID: PMC7185105
- DOI: 10.18632/aging.102977
Down-regulation of AMPK signaling pathway rescues hearing loss in TFB1 transgenic mice and delays age-related hearing loss
Abstract
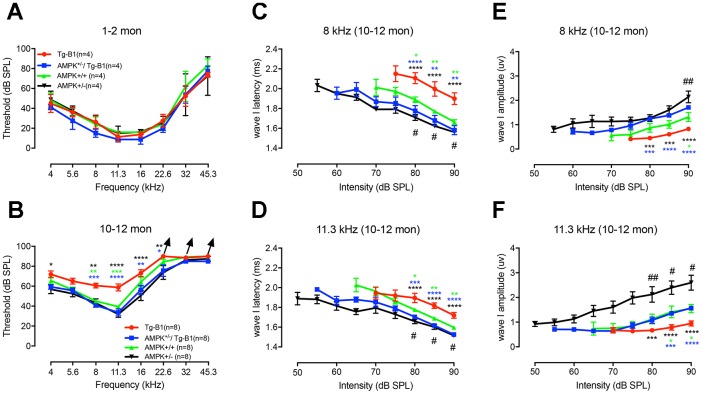
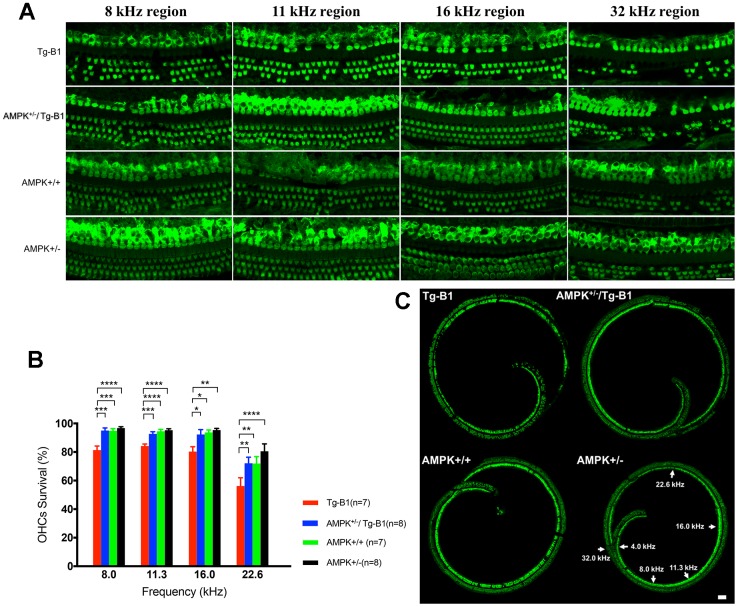
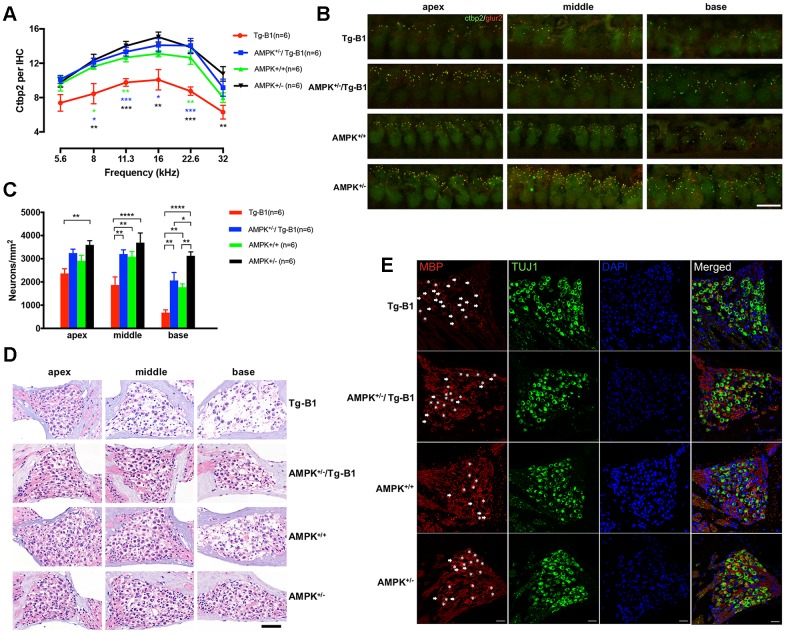
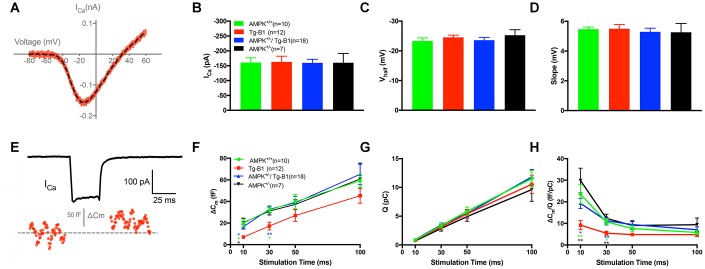
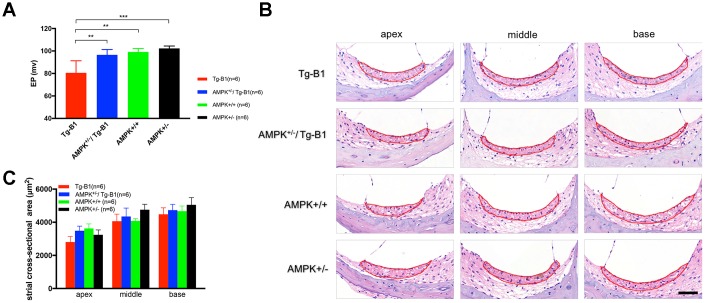
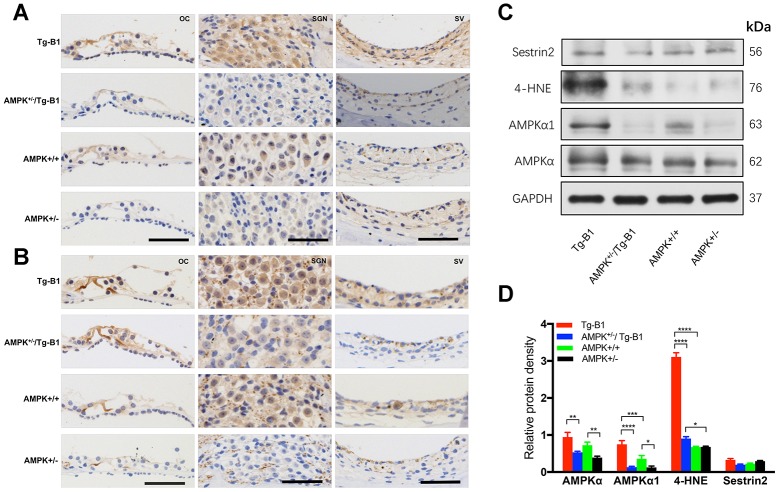
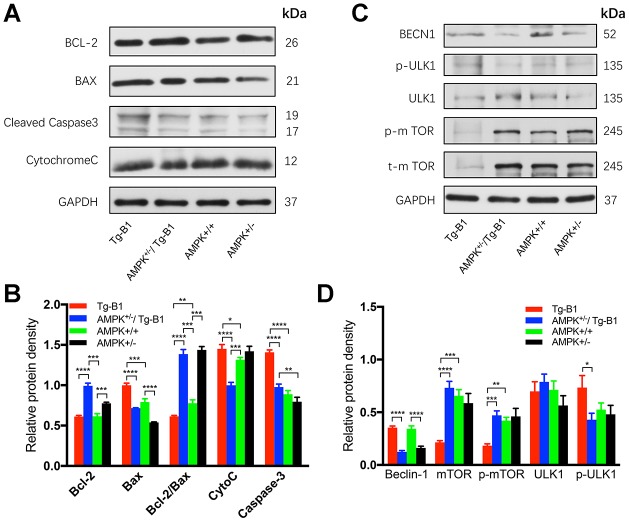
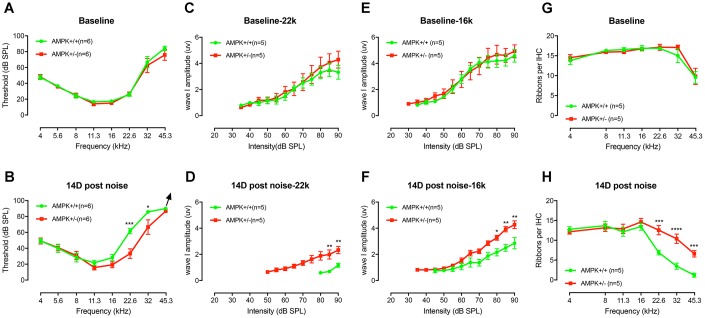
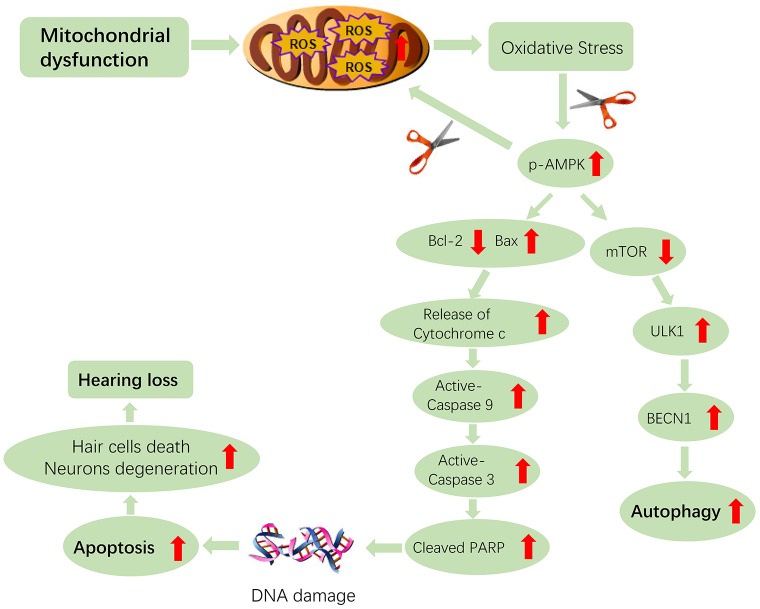
AMP-activated protein kinase (AMPK) integrates the regulation of cell growth and metabolism. AMPK activation occurs in response to cellular energy decline and mitochondrial dysfunction triggered by reactive oxygen species (ROS). In aged Tg-mtTFB1 mice, a mitochondrial deafness mouse model, hearing loss is accompanied with cochlear pathology including reduced endocochlear potential (EP) and loss of spiral ganglion neurons (SGN), inner hair cell (IHC) synapses and outer hair cells (OHC). Accumulated ROS and increased apoptosis signaling were also detected in cochlear tissues, accompanied by activation of AMPK. To further explore the role of AMPK signaling in the auditory phenotype, we used genetically knocked out AMPKα1 as a rescue to Tg-mtTFB1 mice and observed: improved ABR wave I, EP and IHC function, normal SGNs, IHC synapses morphology and OHC survivals, with decreased ROS, reduced pro-apoptotic signaling (Bax) and increased anti-apoptotic signaling (Bcl-2) in the cochlear tissues, indicating that reduced AMPK attenuated apoptosis via ROS-AMPK-Bcl2 pathway in the cochlea. To conclude, AMPK hyperactivation causes accelerated presbycusis in Tg-mtTFB1 mice by redox imbalance and dysregulation of the apoptosis pathway. The effects of AMPK downregulation on pro-survival function and reduction of oxidative stress indicate AMPK serves as a target to rescue or relieve mitochondrial hearing loss.
Keywords: AMPK; NIHL; ROS; apoptosis; mitochondrial deafness.
Conflict of interest statement
Figures
Similar articles
-
Auditory Pathology in a Transgenic mtTFB1 Mouse Model of Mitochondrial Deafness.Am J Pathol. 2015 Dec;185(12):3132-40. doi: 10.1016/j.ajpath.2015.08.014. Epub 2015 Nov 6. Am J Pathol. 2015. PMID: 26552864 Free PMC article.
-
Noise-Induced Loss of Hair Cells and Cochlear Synaptopathy Are Mediated by the Activation of AMPK.J Neurosci. 2016 Jul 13;36(28):7497-510. doi: 10.1523/JNEUROSCI.0782-16.2016. J Neurosci. 2016. PMID: 27413159 Free PMC article.
-
Wnt Signaling Activates TP53-Induced Glycolysis and Apoptosis Regulator and Protects Against Cisplatin-Induced Spiral Ganglion Neuron Damage in the Mouse Cochlea.Antioxid Redox Signal. 2019 Apr 10;30(11):1389-1410. doi: 10.1089/ars.2017.7288. Epub 2018 May 4. Antioxid Redox Signal. 2019. PMID: 29587485
-
Age-related loss of spiral ganglion neurons.Hear Res. 2010 Jun 1;264(1-2):93-7. doi: 10.1016/j.heares.2009.10.009. Epub 2009 Oct 23. Hear Res. 2010. PMID: 19854255 Free PMC article. Review.
-
The mitochondrion: a perpetrator of acquired hearing loss.Hear Res. 2013 Sep;303:12-9. doi: 10.1016/j.heares.2013.01.006. Epub 2013 Jan 27. Hear Res. 2013. PMID: 23361190 Free PMC article. Review.
Cited by
-
Disruption of Hars2 in Cochlear Hair Cells Causes Progressive Mitochondrial Dysfunction and Hearing Loss in Mice.Front Cell Neurosci. 2021 Dec 15;15:804345. doi: 10.3389/fncel.2021.804345. eCollection 2021. Front Cell Neurosci. 2021. PMID: 34975414 Free PMC article.
-
Alveolar Type II Cell Damage and Nrf2-SOD1 Pathway Downregulation Are Involved in PM2.5-Induced Lung Injury in Rats.Int J Environ Res Public Health. 2022 Oct 8;19(19):12893. doi: 10.3390/ijerph191912893. Int J Environ Res Public Health. 2022. PMID: 36232201 Free PMC article.
-
DMXL2 Is Required for Endocytosis and Recycling of Synaptic Vesicles in Auditory Hair Cells.J Neurosci. 2024 Sep 18;44(38):e1405232024. doi: 10.1523/JNEUROSCI.1405-23.2024. J Neurosci. 2024. PMID: 39147590 Free PMC article.
-
Aligned Organization of Synapses and Mitochondria in Auditory Hair Cells.Neurosci Bull. 2022 Mar;38(3):235-248. doi: 10.1007/s12264-021-00801-w. Epub 2021 Nov 27. Neurosci Bull. 2022. PMID: 34837647 Free PMC article.
-
Involvement of BK Channels and Ryanodine Receptors in Salicylate-induced Tinnitus.Mol Neurobiol. 2024 Oct 14. doi: 10.1007/s12035-024-04533-6. Online ahead of print. Mol Neurobiol. 2024. PMID: 39397241
References
-
- Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996; 271:27879–87. 10.1074/jbc.271.44.27879 - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
