

Avoiding Drug Resistance by Substrate Envelope-Guided Design: Toward Potent and Robust HCV NS3/4A Protease Inhibitors
- PMID: 32234812
- PMCID: PMC7157764
- DOI: 10.1128/mBio.00172-20
Avoiding Drug Resistance by Substrate Envelope-Guided Design: Toward Potent and Robust HCV NS3/4A Protease Inhibitors
Abstract
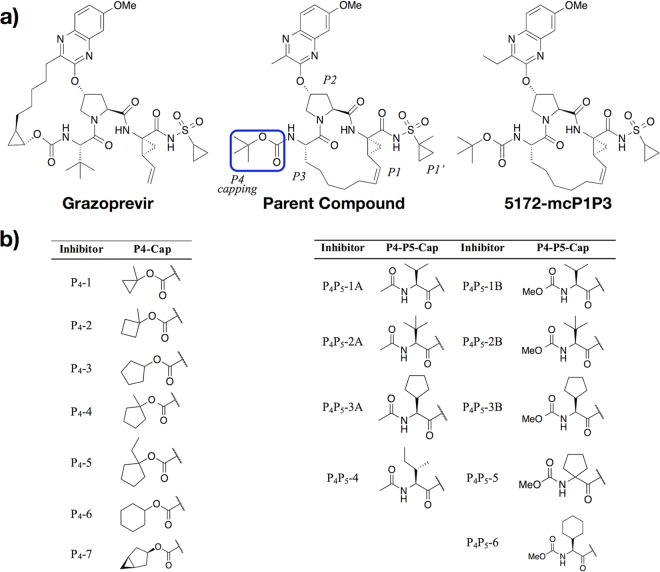
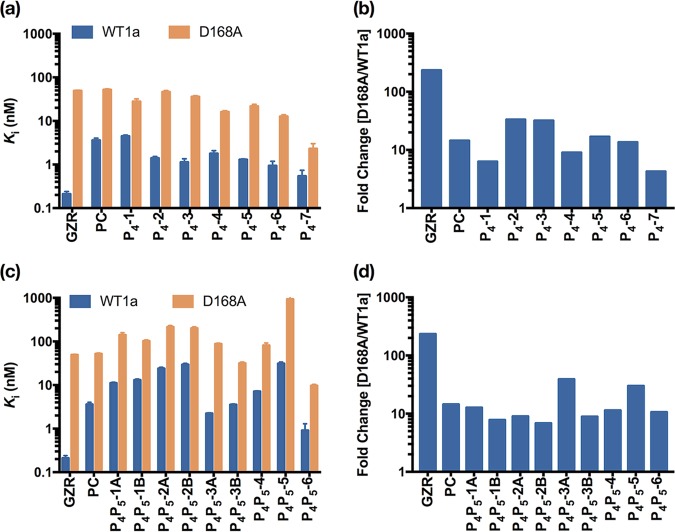
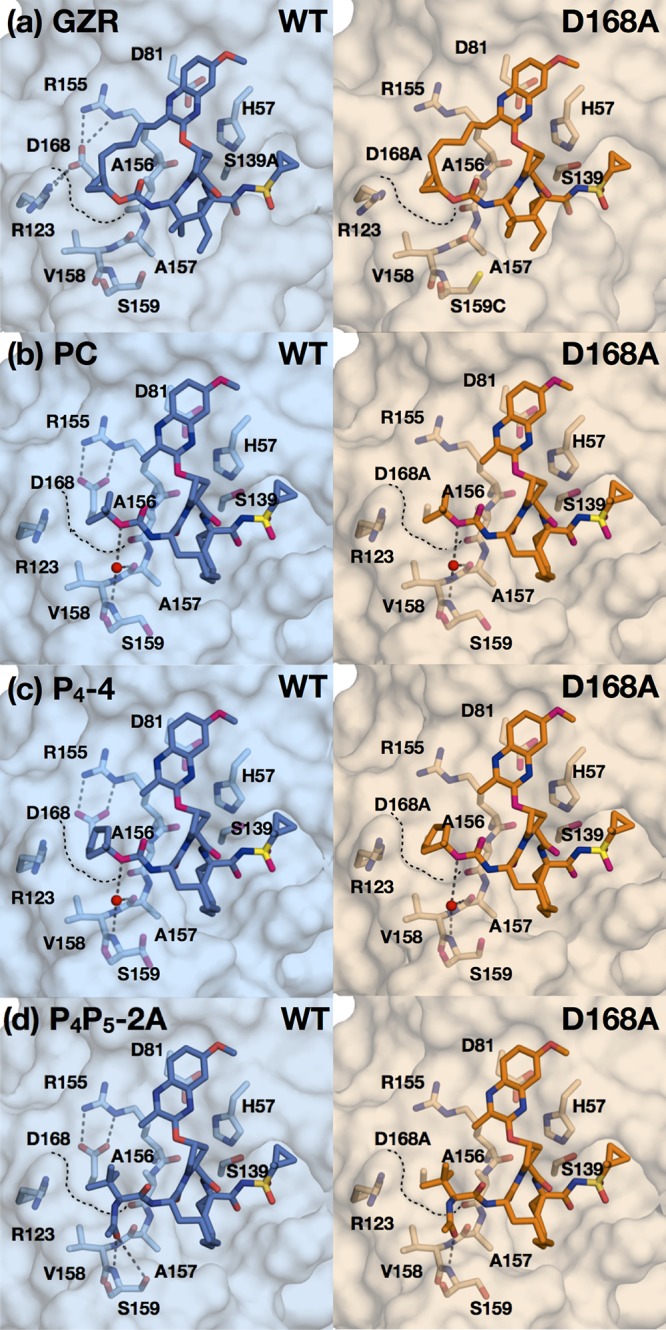
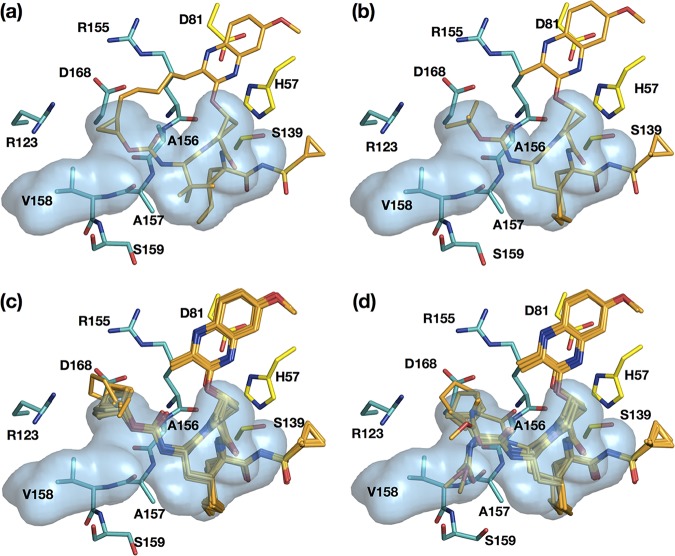
Hepatitis C virus (HCV) infects millions of people worldwide, causing chronic liver disease that can lead to cirrhosis, hepatocellular carcinoma, and liver transplant. In the last several years, the advent of direct-acting antivirals, including NS3/4A protease inhibitors (PIs), has remarkably improved treatment outcomes of HCV-infected patients. However, selection of resistance-associated substitutions and polymorphisms among genotypes can lead to drug resistance and in some cases treatment failure. A proactive strategy to combat resistance is to constrain PIs within evolutionarily conserved regions in the protease active site. Designing PIs using the substrate envelope is a rational strategy to decrease the susceptibility to resistance by using the constraints of substrate recognition. We successfully designed two series of HCV NS3/4A PIs to leverage unexploited areas in the substrate envelope to improve potency, specifically against resistance-associated substitutions at D168. Our design strategy achieved better resistance profiles over both the FDA-approved NS3/4A PI grazoprevir and the parent compound against the clinically relevant D168A substitution. Crystallographic structural analysis and inhibition assays confirmed that optimally filling the substrate envelope is critical to improve inhibitor potency while avoiding resistance. Specifically, inhibitors that enhanced hydrophobic packing in the S4 pocket and avoided an energetically frustrated pocket performed the best. Thus, the HCV substrate envelope proved to be a powerful tool to design robust PIs, offering a strategy that can be translated to other targets for rational design of inhibitors with improved potency and resistance profiles.IMPORTANCE Despite significant progress, hepatitis C virus (HCV) continues to be a major health problem with millions of people infected worldwide and thousands dying annually due to resulting complications. Recent antiviral combinations can achieve >95% cure, but late diagnosis, low access to treatment, and treatment failure due to drug resistance continue to be roadblocks against eradication of the virus. We report the rational design of two series of HCV NS3/4A protease inhibitors with improved resistance profiles by exploiting evolutionarily constrained regions of the active site using the substrate envelope model. Optimally filling the S4 pocket is critical to avoid resistance and improve potency. Our results provide drug design strategies to avoid resistance that are applicable to other quickly evolving viral drug targets.
Keywords: X-ray crystallography; drug design; drug resistance mechanisms; hepatitis C virus; structural biology.
Copyright © 2020 Matthew et al.
Figures
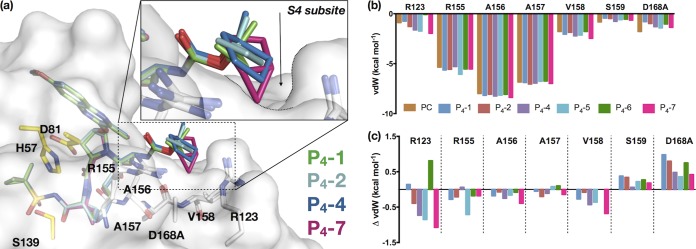
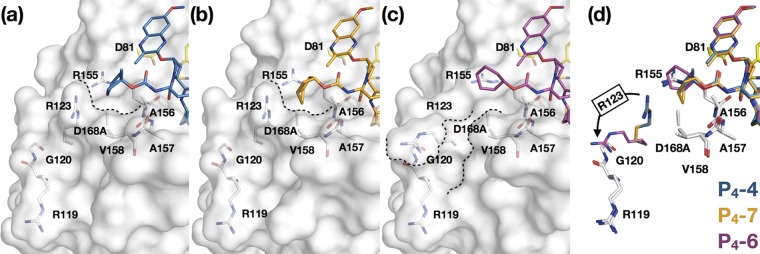
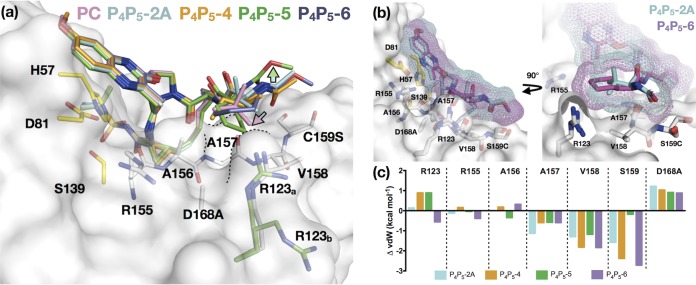
Similar articles
-
Understanding the structural and energetic basis of inhibitor and substrate bound to the full-length NS3/4A: insights from molecular dynamics simulation, binding free energy calculation and network analysis.Mol Biosyst. 2012 Oct;8(10):2753-65. doi: 10.1039/c2mb25157d. Mol Biosyst. 2012. PMID: 22833015
-
Molecular Mechanism of Resistance in a Clinically Significant Double-Mutant Variant of HCV NS3/4A Protease.Structure. 2018 Oct 2;26(10):1360-1372.e5. doi: 10.1016/j.str.2018.07.004. Epub 2018 Aug 23. Structure. 2018. PMID: 30146168 Free PMC article.
-
Computational study on the drug resistance mechanism against HCV NS3/4A protease inhibitors vaniprevir and MK-5172 by the combination use of molecular dynamics simulation, residue interaction network, and substrate envelope analysis.J Chem Inf Model. 2014 Feb 24;54(2):621-33. doi: 10.1021/ci400060j. Epub 2013 Jun 28. J Chem Inf Model. 2014. PMID: 23745769
-
Resistance outside the substrate envelope: hepatitis C NS3/4A protease inhibitors.Crit Rev Biochem Mol Biol. 2019 Feb;54(1):11-26. doi: 10.1080/10409238.2019.1568962. Epub 2019 Mar 1. Crit Rev Biochem Mol Biol. 2019. PMID: 30821513 Free PMC article. Review.
-
Hepatitis C virus resistance to protease inhibitors.J Hepatol. 2011 Jul;55(1):192-206. doi: 10.1016/j.jhep.2011.01.011. Epub 2011 Feb 1. J Hepatol. 2011. PMID: 21284949 Review.
Cited by
-
N4-Hydroxycytidine/molnupiravir inhibits RNA virus-induced encephalitis by producing less fit mutated viruses.PLoS Pathog. 2024 Sep 30;20(9):e1012574. doi: 10.1371/journal.ppat.1012574. eCollection 2024 Sep. PLoS Pathog. 2024. PMID: 39348391 Free PMC article.
-
Viral proteases as therapeutic targets.Mol Aspects Med. 2022 Dec;88:101159. doi: 10.1016/j.mam.2022.101159. Epub 2022 Nov 29. Mol Aspects Med. 2022. PMID: 36459838 Free PMC article. Review.
-
Drug Design Strategies to Avoid Resistance in Direct-Acting Antivirals and Beyond.Chem Rev. 2021 Mar 24;121(6):3238-3270. doi: 10.1021/acs.chemrev.0c00648. Epub 2021 Jan 7. Chem Rev. 2021. PMID: 33410674 Free PMC article. Review.
-
Report of the National Institutes of Health SARS-CoV-2 Antiviral Therapeutics Summit.J Infect Dis. 2021 Jul 15;224(Supplement_1):S1-S21. doi: 10.1093/infdis/jiab305. J Infect Dis. 2021. PMID: 34111271 Free PMC article.
-
Defining the substrate envelope of SARS-CoV-2 main protease to predict and avoid drug resistance.Nat Commun. 2022 Jun 21;13(1):3556. doi: 10.1038/s41467-022-31210-w. Nat Commun. 2022. PMID: 35729165 Free PMC article.
References
-
- WHO. 2018. Hepatitis C fact sheet (updated July 2018). https://www.who.int/en/news-room/fact-sheets/detail/hepatitis-c.
-
- Ghany MG, Morgan TR, AASLD-IDSA Hepatitis C Guidance Panel. 2020. Hepatitis C guidance 2019 update: American Association for the Study of Liver Diseases-Infectious Diseases Society of America recommendations for testing, managing, and treating hepatitis C virus infection. Hepatology 71:686–721. doi:10.1002/hep.31060. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
