

Neurofibromin Is an Estrogen Receptor-α Transcriptional Co-repressor in Breast Cancer
- PMID: 32142667
- PMCID: PMC7286719
- DOI: 10.1016/j.ccell.2020.02.003
Neurofibromin Is an Estrogen Receptor-α Transcriptional Co-repressor in Breast Cancer
Abstract
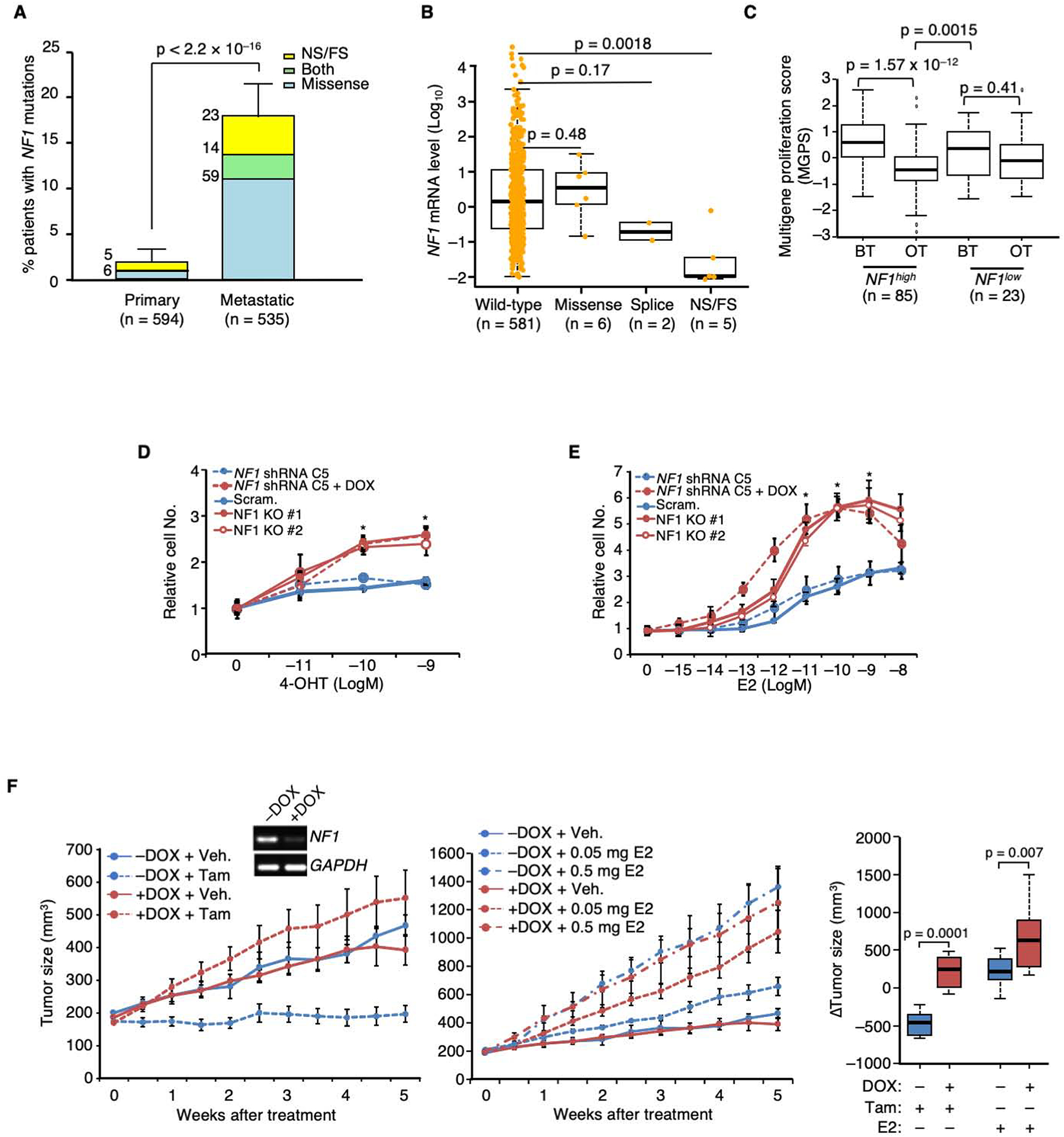
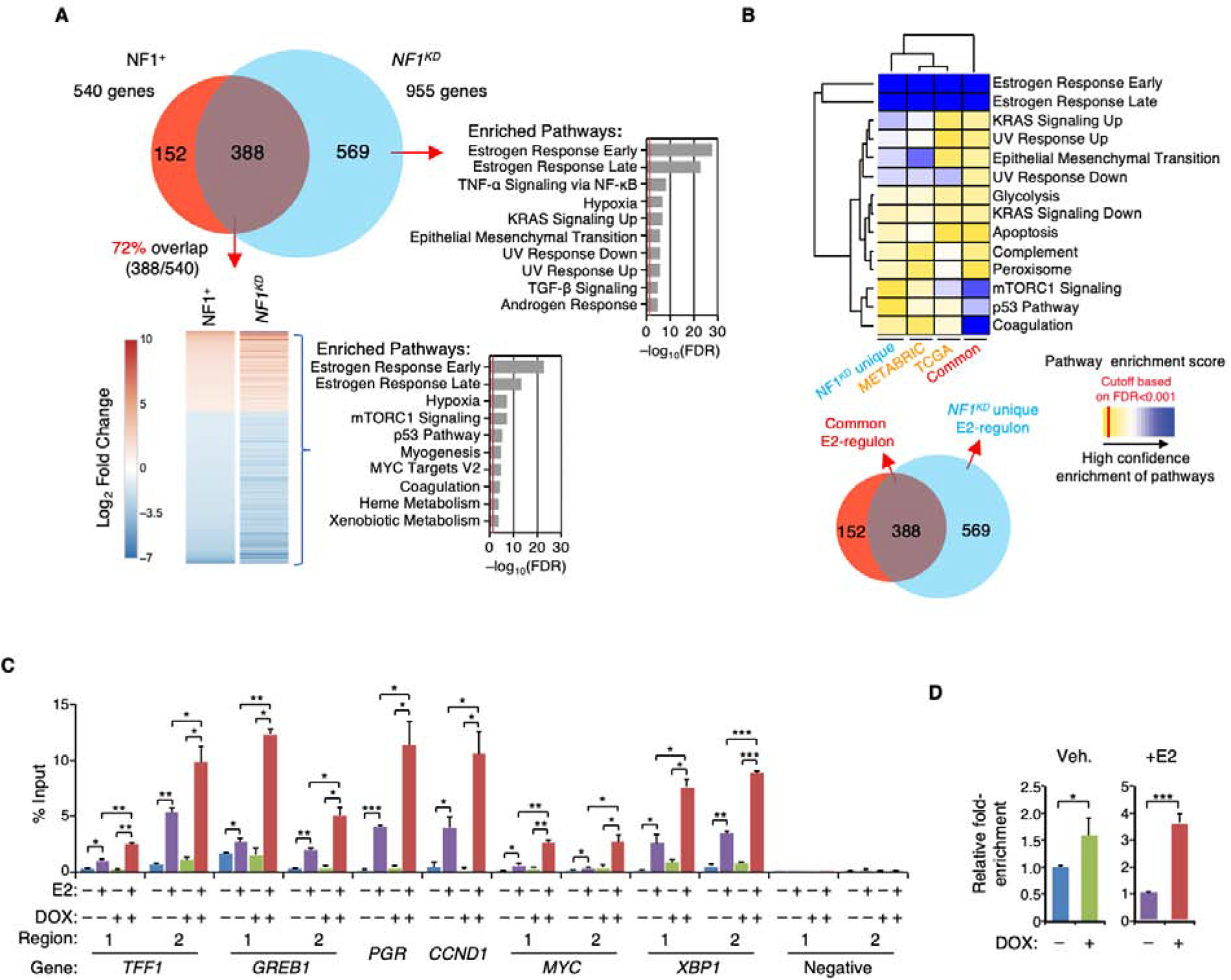
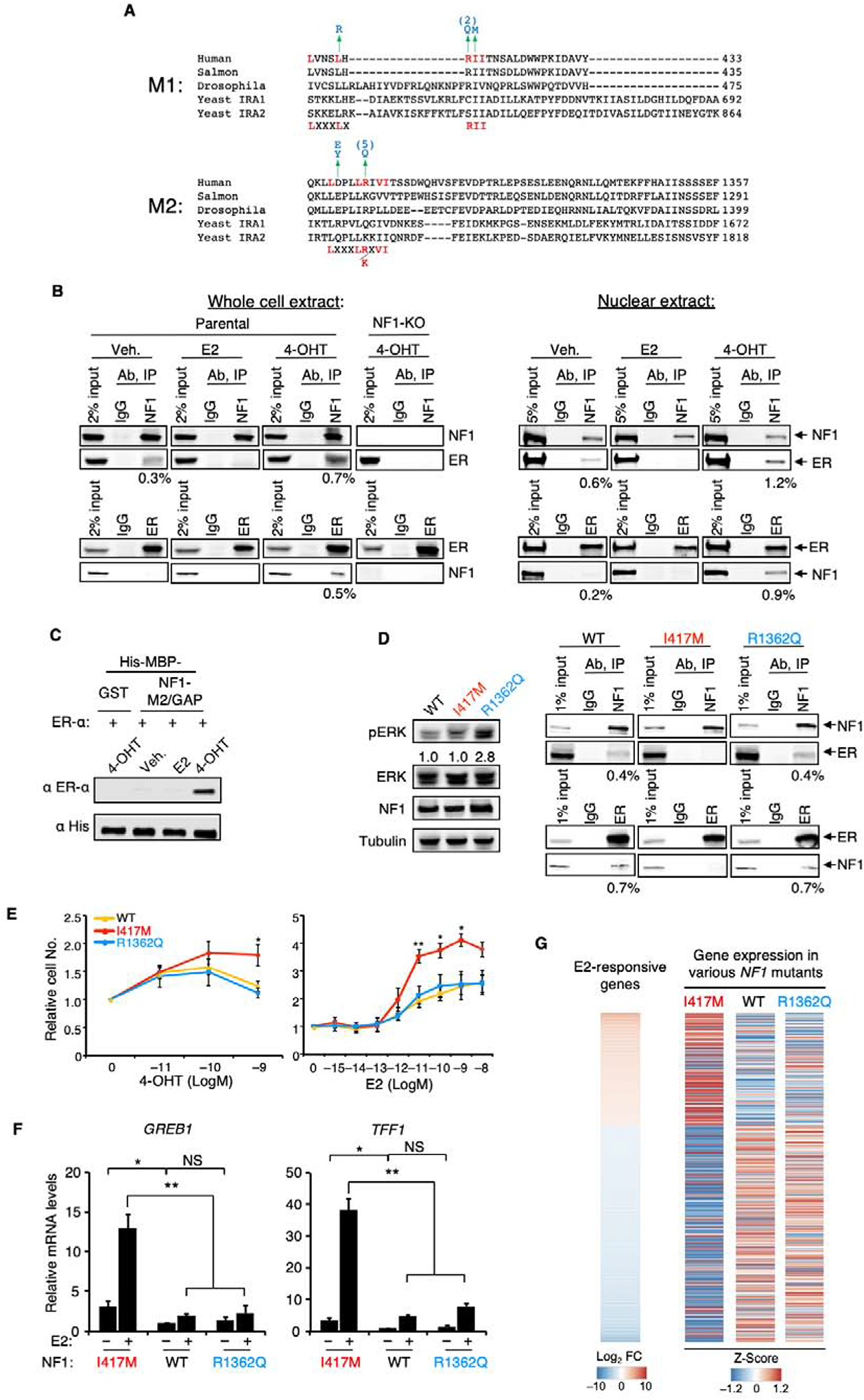
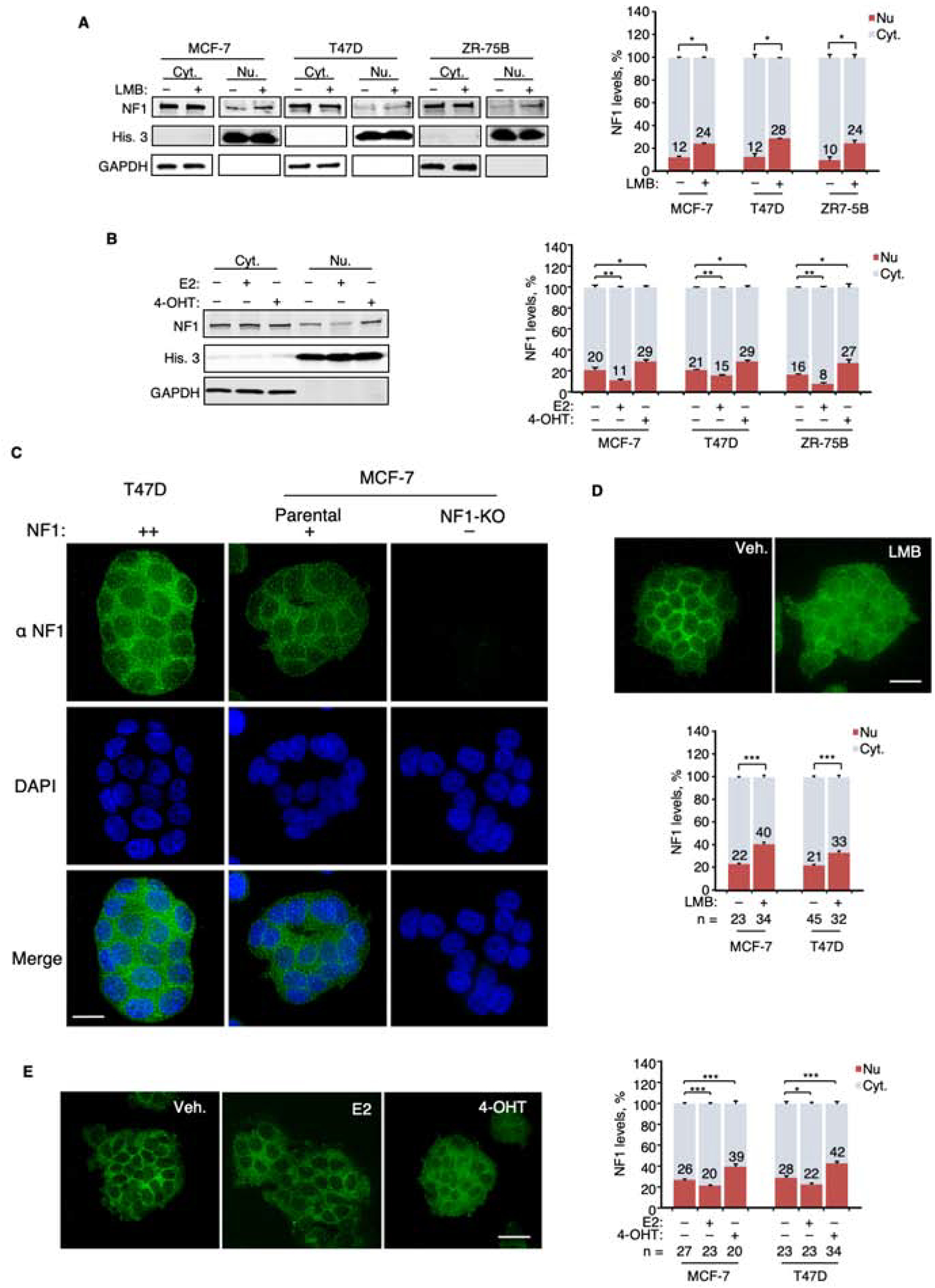
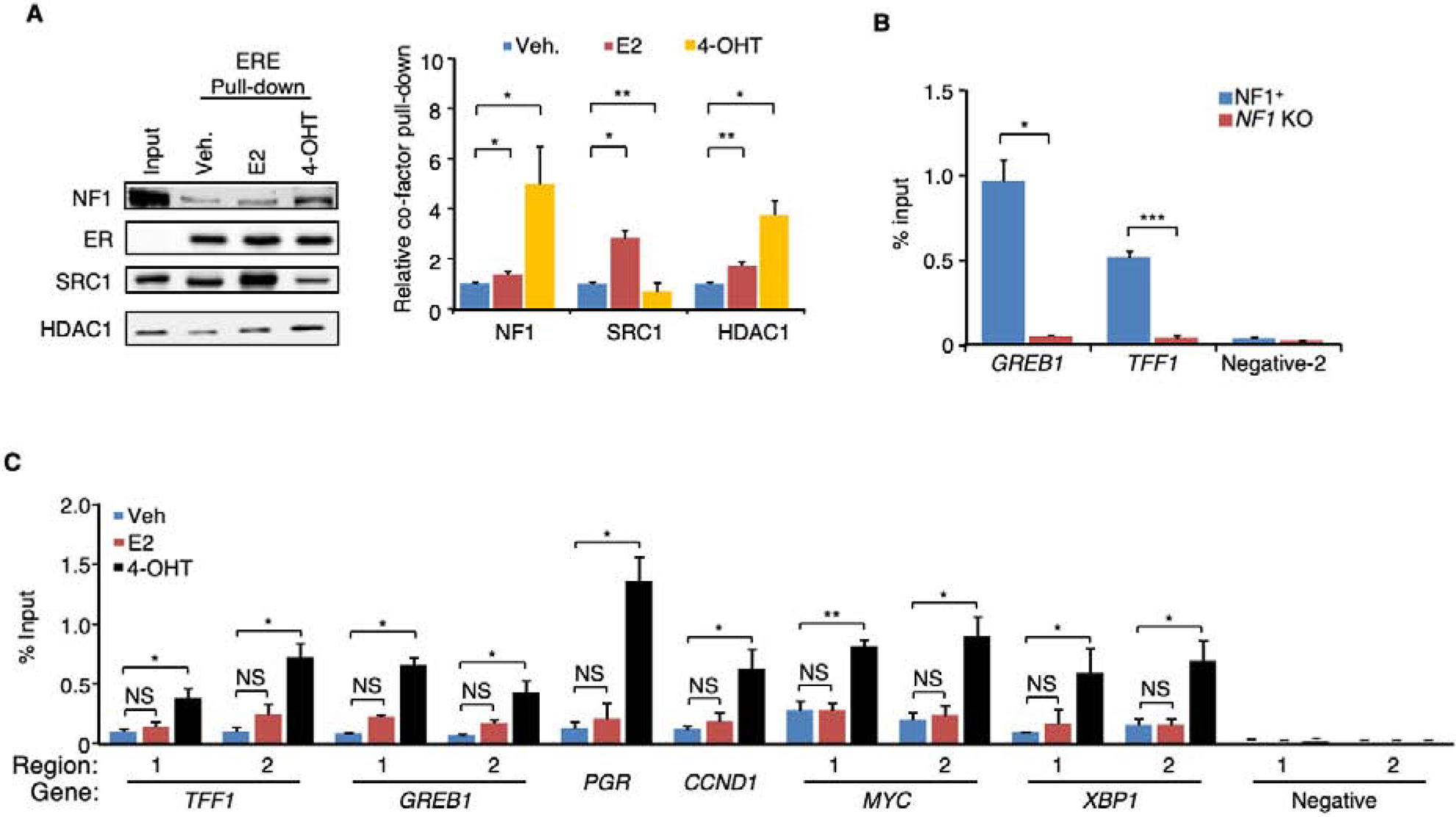
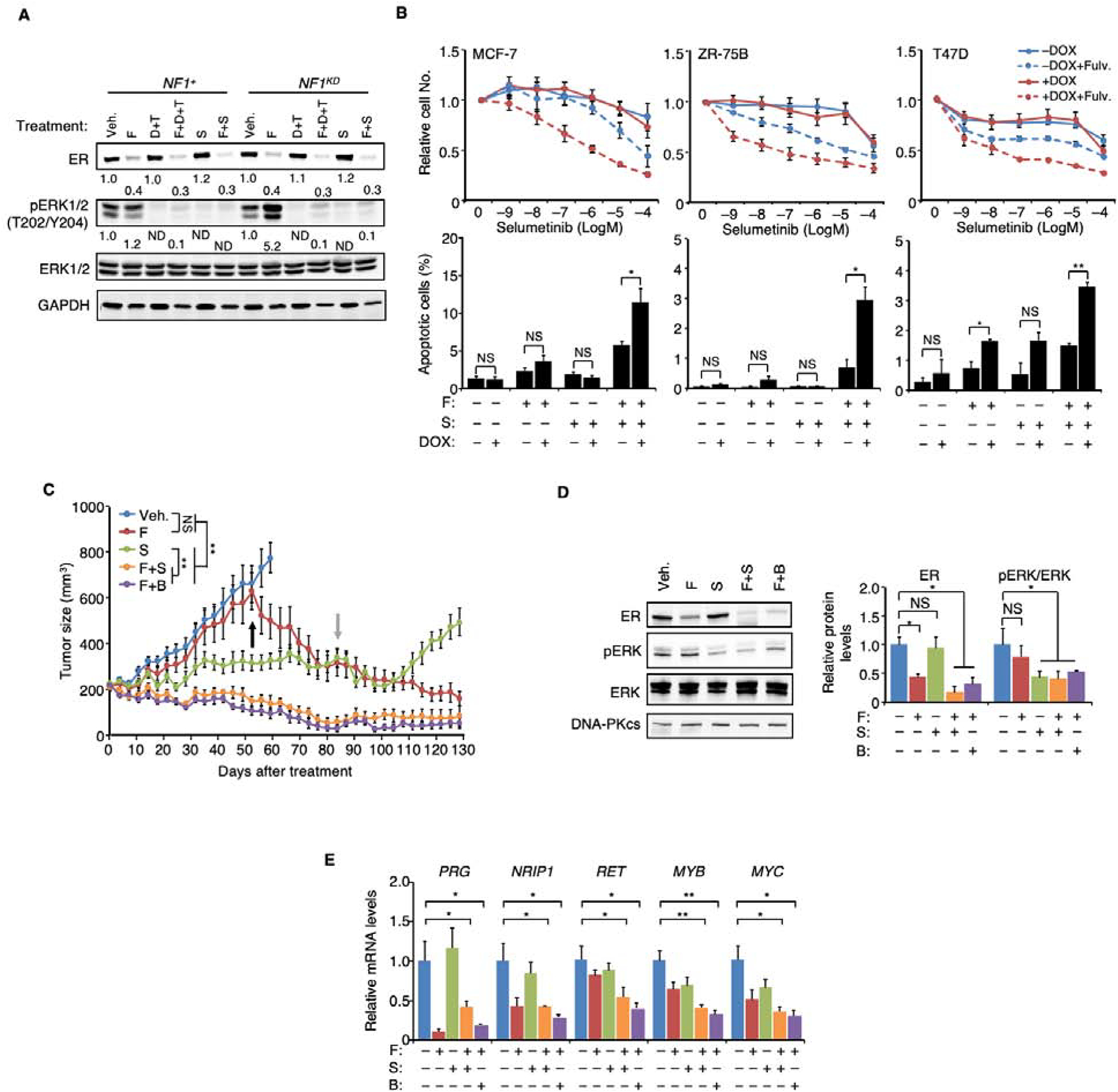
We report that neurofibromin, a tumor suppressor and Ras-GAP (GTPase-activating protein), is also an estrogen receptor-α (ER) transcriptional co-repressor through leucine/isoleucine-rich motifs that are functionally independent of GAP activity. GAP activity, in turn, does not affect ER binding. Consequently, neurofibromin depletion causes estradiol hypersensitivity and tamoxifen agonism, explaining the poor prognosis associated with neurofibromin loss in endocrine therapy-treated ER+ breast cancer. Neurofibromin-deficient ER+ breast cancer cells initially retain sensitivity to selective ER degraders (SERDs). However, Ras activation does play a role in acquired SERD resistance, which can be reversed upon MEK inhibitor addition, and SERD/MEK inhibitor combinations induce tumor regression. Thus, neurofibromin is a dual repressor for both Ras and ER signaling, and co-targeting may treat neurofibromin-deficient ER+ breast tumors.
Keywords: Drosophila; GTPase; NF1; RAS; breast cancer; co-regulator; endocrine therapy; estrogen receptor; neurofibromatosis; yeast.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests K.C.B. and R.B.L. are stockholders and employees of Guardant Health, Inc., C.E.F. discloses an equity position in Coactigon, Inc., and M.J.E. received consulting fees from Abbvie, Sermonix, Pfizer, AstraZeneca, Celgene, NanoString, Puma, and Novartis, and is an equity stockholder, consultant, and Board Director member of BioClassifier, and inventor on a patent for the Breast PAM50 assay.
Figures
Similar articles
-
Efficacy of SERD/SERM Hybrid-CDK4/6 Inhibitor Combinations in Models of Endocrine Therapy-Resistant Breast Cancer.Clin Cancer Res. 2015 Nov 15;21(22):5121-5130. doi: 10.1158/1078-0432.CCR-15-0360. Epub 2015 May 19. Clin Cancer Res. 2015. PMID: 25991817 Free PMC article.
-
High estrogen receptor alpha activation confers resistance to estrogen deprivation and is required for therapeutic response to estrogen in breast cancer.Oncogene. 2021 May;40(19):3408-3421. doi: 10.1038/s41388-021-01782-w. Epub 2021 Apr 19. Oncogene. 2021. PMID: 33875787 Free PMC article.
-
NF1 deficiency drives metabolic reprogramming in ER+ breast cancer.Mol Metab. 2024 Feb;80:101876. doi: 10.1016/j.molmet.2024.101876. Epub 2024 Jan 10. Mol Metab. 2024. PMID: 38216123 Free PMC article.
-
Neurofibromatosis 1: closing the GAP between mice and men.Curr Opin Genet Dev. 2003 Feb;13(1):20-7. doi: 10.1016/s0959-437x(02)00015-1. Curr Opin Genet Dev. 2003. PMID: 12573431 Review.
-
Role of estrogen receptor alpha transcriptional coregulators in tamoxifen resistance in breast cancer.Maturitas. 2006 Jul 20;54(4):342-51. doi: 10.1016/j.maturitas.2006.06.003. Epub 2006 Jul 5. Maturitas. 2006. PMID: 16822624 Review.
Cited by
-
Genomic Alterations Associated with Estrogen Receptor Pathway Activity in Metastatic Breast Cancer Have a Differential Impact on Downstream ER Signaling.Cancers (Basel). 2023 Sep 4;15(17):4416. doi: 10.3390/cancers15174416. Cancers (Basel). 2023. PMID: 37686693 Free PMC article.
-
Genetic Events and Signaling Mechanisms Underlying Schwann Cell Fate in Development and Cancer.Neurosurgery. 2021 Jan 13;88(2):234-245. doi: 10.1093/neuros/nyaa455. Neurosurgery. 2021. PMID: 33094349 Free PMC article. Review.
-
RAS and beyond: the many faces of the neurofibromatosis type 1 protein.Dis Model Mech. 2022 Feb 1;15(2):dmm049362. doi: 10.1242/dmm.049362. Epub 2022 Feb 21. Dis Model Mech. 2022. PMID: 35188187 Free PMC article.
-
Increased serum concentrations of estrogen-induced growth factors Midkine and FGF2 in NF1 patients with plexiform neurofibroma.Am J Transl Res. 2022 May 15;14(5):3180-3188. eCollection 2022. Am J Transl Res. 2022. PMID: 35702135 Free PMC article.
-
Acquired mutations and transcriptional remodeling in long-term estrogen-deprived locoregional breast cancer recurrences.Breast Cancer Res. 2021 Jan 6;23(1):1. doi: 10.1186/s13058-020-01379-3. Breast Cancer Res. 2021. PMID: 33407744 Free PMC article.
References
-
- Anbalagan M, and Rowan BG (2015). Estrogen receptor alpha phosphorylation and its functional impact in human breast cancer. Mol Cell Endocrinol 418 Pt 3, 264–272. - PubMed
-
- Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, and Collins F (1990). The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 63, 851–859. - PubMed
-
- Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, Soria JC, Dien AT, Adnani Y, Kamal M, et al. (2019). Genomic characterization of metastatic breast cancers. Nature 569, 560–564. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
