

Chromosome-Level Assembly of the Caenorhabditis remanei Genome Reveals Conserved Patterns of Nematode Genome Organization
- PMID: 32111628
- PMCID: PMC7153949
- DOI: 10.1534/genetics.119.303018
Chromosome-Level Assembly of the Caenorhabditis remanei Genome Reveals Conserved Patterns of Nematode Genome Organization
Abstract
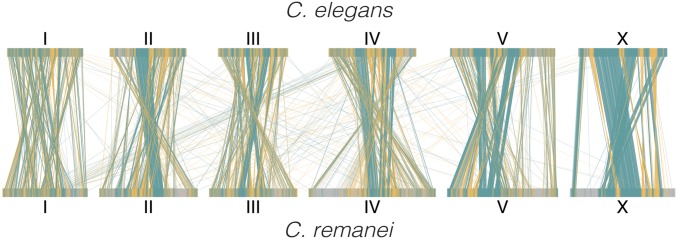
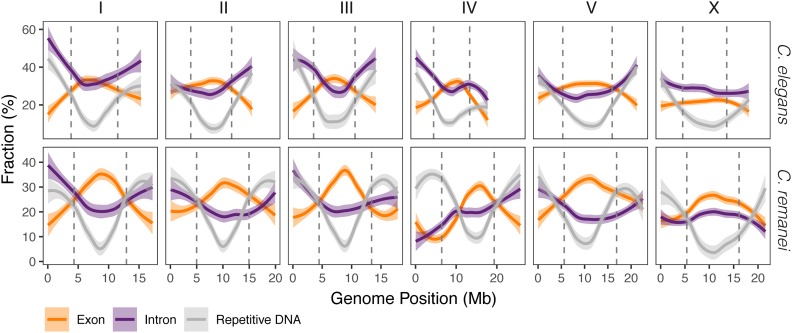
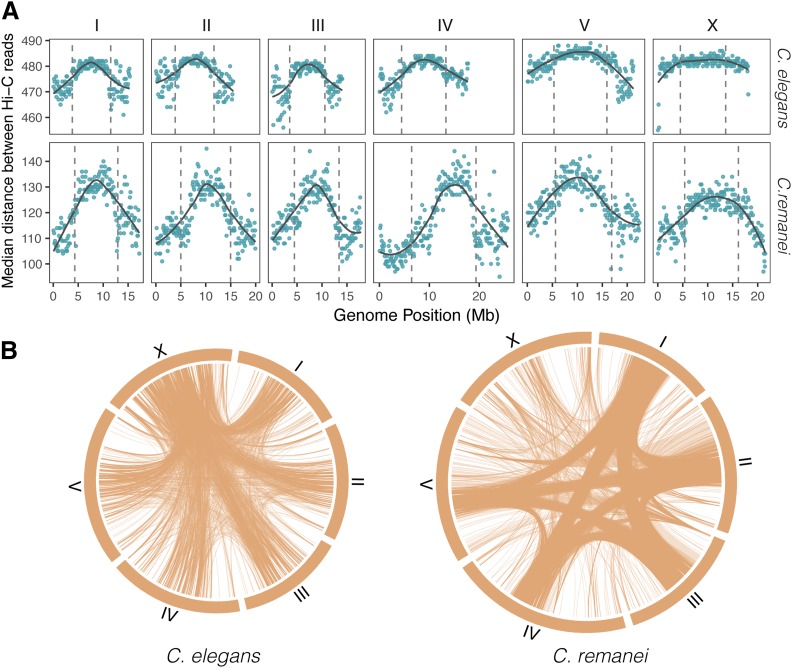
The nematode Caenorhabditis elegans is one of the key model systems in biology, including possessing the first fully assembled animal genome. Whereas C. elegans is a self-reproducing hermaphrodite with fairly limited within-population variation, its relative C. remanei is an outcrossing species with much more extensive genetic variation, making it an ideal parallel model system for evolutionary genetic investigations. Here, we greatly improve on previous assemblies by generating a chromosome-level assembly of the entire C. remanei genome (124.8 Mb of total size) using long-read sequencing and chromatin conformation capture data. Like other fully assembled genomes in the genus, we find that the C. remanei genome displays a high degree of synteny with C. elegans despite multiple within-chromosome rearrangements. Both genomes have high gene density in central regions of chromosomes relative to chromosome ends and the opposite pattern for the accumulation of repetitive elements. C. elegans and C. remanei also show similar patterns of interchromosome interactions, with the central regions of chromosomes appearing to interact with one another more than the distal ends. The new C. remanei genome presented here greatly augments the use of the Caenorhabditis as a platform for comparative genomics and serves as a basis for molecular population genetics within this highly diverse species.
Keywords: Caenorhabditis elegans; Caenorhabditis remanei; chromosome-level assembly; comparative genomics.
Copyright © 2020 by the Genetics Society of America.
Figures
Similar articles
-
Reproductive Mode and the Evolution of Genome Size and Structure in Caenorhabditis Nematodes.PLoS Genet. 2015 Jun 26;11(6):e1005323. doi: 10.1371/journal.pgen.1005323. eCollection 2015 Jun. PLoS Genet. 2015. PMID: 26114425 Free PMC article.
-
Global population genetic structure of Caenorhabditis remanei reveals incipient speciation.Genetics. 2012 Aug;191(4):1257-69. doi: 10.1534/genetics.112.140418. Epub 2012 May 29. Genetics. 2012. PMID: 22649079 Free PMC article.
-
Comparison of C. elegans and C. briggsae genome sequences reveals extensive conservation of chromosome organization and synteny.PLoS Biol. 2007 Jul;5(7):e167. doi: 10.1371/journal.pbio.0050167. Epub 2007 Jul 3. PLoS Biol. 2007. PMID: 17608563 Free PMC article.
-
Genome sequence of the nematode C. elegans: a platform for investigating biology.Science. 1998 Dec 11;282(5396):2012-8. doi: 10.1126/science.282.5396.2012. Science. 1998. PMID: 9851916 Review.
-
Comparative genomics in C. elegans, C. briggsae, and other Caenorhabditis species.Methods Mol Biol. 2006;351:13-29. doi: 10.1385/1-59745-151-7:13. Methods Mol Biol. 2006. PMID: 16988423 Review.
Cited by
-
Genome Size Changes by Duplication, Divergence, and Insertion in Caenorhabditis Worms.Mol Biol Evol. 2023 Mar 4;40(3):msad039. doi: 10.1093/molbev/msad039. Mol Biol Evol. 2023. PMID: 36807460 Free PMC article.
-
A telomere-to-telomere assembly of Oscheius tipulae and the evolution of rhabditid nematode chromosomes.G3 (Bethesda). 2021 Jan 18;11(1):jkaa020. doi: 10.1093/g3journal/jkaa020. G3 (Bethesda). 2021. PMID: 33561231 Free PMC article.
-
Ancient diversity in host-parasite interaction genes in a model parasitic nematode.Nat Commun. 2023 Nov 27;14(1):7776. doi: 10.1038/s41467-023-43556-w. Nat Commun. 2023. PMID: 38012132 Free PMC article.
-
Novel and improved Caenorhabditis briggsae gene models generated by community curation.bioRxiv [Preprint]. 2023 May 18:2023.05.16.541014. doi: 10.1101/2023.05.16.541014. bioRxiv. 2023. Update in: BMC Genomics. 2023 Aug 25;24(1):486. doi: 10.1186/s12864-023-09582-0. PMID: 37292880 Free PMC article. Updated. Preprint.
-
Experimental evolution of hybrid populations to identify Dobzhansky-Muller incompatibility loci.Ecol Evol. 2024 Feb 8;14(2):e10972. doi: 10.1002/ece3.10972. eCollection 2024 Feb. Ecol Evol. 2024. PMID: 38333096 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
