

Recounting the FANTOM CAGE-Associated Transcriptome
- PMID: 32079618
- PMCID: PMC7397872
- DOI: 10.1101/gr.254656.119
Recounting the FANTOM CAGE-Associated Transcriptome
Abstract
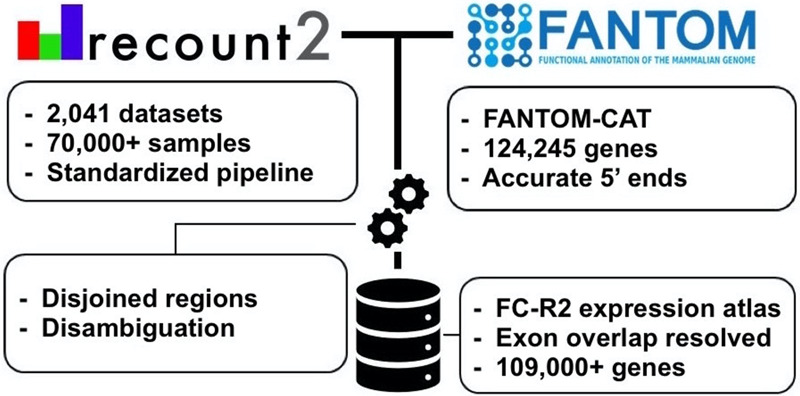
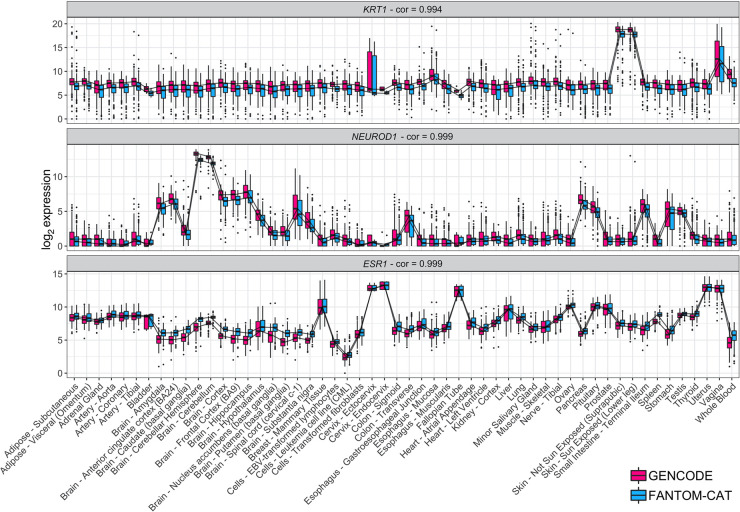
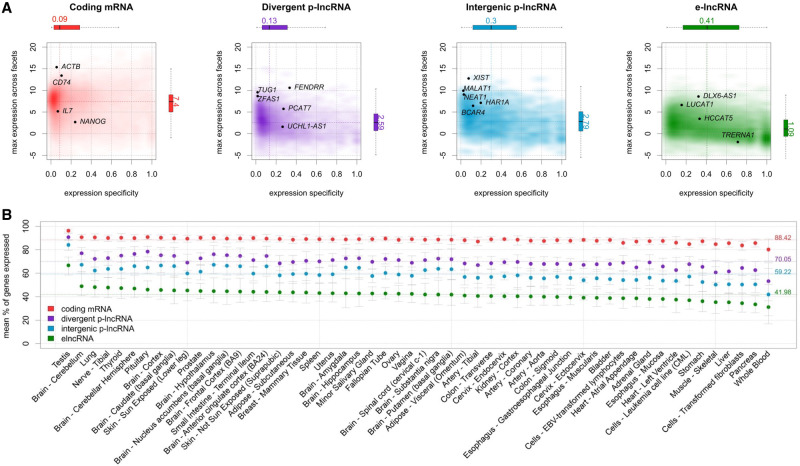
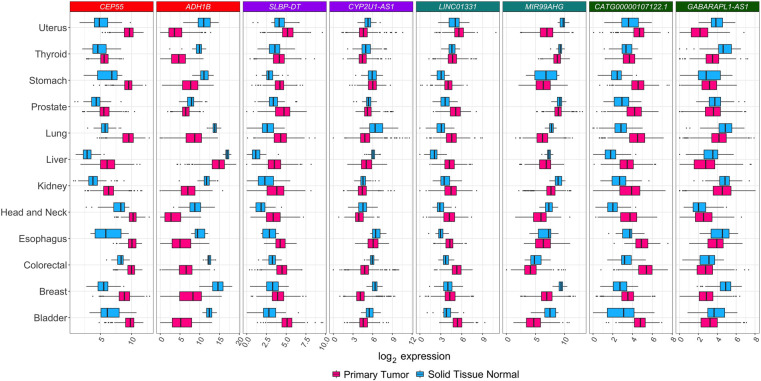
Long noncoding RNAs (lncRNAs) have emerged as key coordinators of biological and cellular processes. Characterizing lncRNA expression across cells and tissues is key to understanding their role in determining phenotypes, including human diseases. We present here FC-R2, a comprehensive expression atlas across a broadly defined human transcriptome, inclusive of over 109,000 coding and noncoding genes, as described in the FANTOM CAGE-Associated Transcriptome (FANTOM-CAT) study. This atlas greatly extends the gene annotation used in the original recount2 resource. We demonstrate the utility of the FC-R2 atlas by reproducing key findings from published large studies and by generating new results across normal and diseased human samples. In particular, we (a) identify tissue-specific transcription profiles for distinct classes of coding and noncoding genes, (b) perform differential expression analysis across thirteen cancer types, identifying novel noncoding genes potentially involved in tumor pathogenesis and progression, and (c) confirm the prognostic value for several enhancer lncRNAs expression in cancer. Our resource is instrumental for the systematic molecular characterization of lncRNA by the FANTOM6 Consortium. In conclusion, comprised of over 70,000 samples, the FC-R2 atlas will empower other researchers to investigate functions and biological roles of both known coding genes and novel lncRNAs.
© 2020 Imada et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
An atlas of human long non-coding RNAs with accurate 5' ends.Nature. 2017 Mar 9;543(7644):199-204. doi: 10.1038/nature21374. Epub 2017 Mar 1. Nature. 2017. PMID: 28241135 Free PMC article.
-
FANTOM enters 20th year: expansion of transcriptomic atlases and functional annotation of non-coding RNAs.Nucleic Acids Res. 2021 Jan 8;49(D1):D892-D898. doi: 10.1093/nar/gkaa1054. Nucleic Acids Res. 2021. PMID: 33211864 Free PMC article.
-
Expression Specificity of Disease-Associated lncRNAs: Toward Personalized Medicine.Curr Top Microbiol Immunol. 2016;394:237-58. doi: 10.1007/82_2015_464. Curr Top Microbiol Immunol. 2016. PMID: 26318140 Review.
-
Long noncoding RNA repertoire in chicken liver and adipose tissue.Genet Sel Evol. 2017 Jan 10;49(1):6. doi: 10.1186/s12711-016-0275-0. Genet Sel Evol. 2017. PMID: 28073357 Free PMC article.
-
Databases for lncRNAs: a comparative evaluation of emerging tools.RNA. 2014 Nov;20(11):1655-65. doi: 10.1261/rna.044040.113. RNA. 2014. PMID: 25323317 Free PMC article. Review.
Cited by
-
The annotation of GBA1 has been concealed by its protein-coding pseudogene GBAP1.Sci Adv. 2024 Jun 28;10(26):eadk1296. doi: 10.1126/sciadv.adk1296. Epub 2024 Jun 26. Sci Adv. 2024. PMID: 38924406 Free PMC article.
-
Loss of heterozygosity impacts MHC expression on the immune microenvironment in CDK12-mutated prostate cancer.Mol Cytogenet. 2024 May 4;17(1):11. doi: 10.1186/s13039-024-00680-6. Mol Cytogenet. 2024. PMID: 38704603 Free PMC article.
-
Pan-cancer genomic analysis shows hemizygous PTEN loss tumors are associated with immune evasion and poor outcome.Sci Rep. 2023 Mar 28;13(1):5049. doi: 10.1038/s41598-023-31759-6. Sci Rep. 2023. PMID: 36977733 Free PMC article.
-
Pan-Cancer Analysis Reveals Functional Similarity of Three lncRNAs across Multiple Tumors.Int J Mol Sci. 2023 Mar 1;24(5):4796. doi: 10.3390/ijms24054796. Int J Mol Sci. 2023. PMID: 36902227 Free PMC article.
-
Identification of androgen response-related lncRNAs in prostate cancer.Prostate. 2023 May;83(6):590-601. doi: 10.1002/pros.24494. Epub 2023 Feb 9. Prostate. 2023. PMID: 36760203 Free PMC article.
References
-
- Arner E, Daub CO, Vitting-Seerup K, Andersson R, Lilje B, Drabløs F, Lennartsson A, Rönnerblad M, Hrydziuszko O, Vitezic M, et al. 2015. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347: 1010–1014. 10.1126/science.1259418 - DOI - PMC - PubMed
-
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B 57: 289–300. 10.1111/j.2517-6161.1995.tb02031.x - DOI
-
- Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. 2019. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47: D1005–D1012. 10.1093/nar/gky1120 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous