

Analyses of non-coding somatic drivers in 2,658 cancer whole genomes
- PMID: 32025015
- PMCID: PMC7054214
- DOI: 10.1038/s41586-020-1965-x
Analyses of non-coding somatic drivers in 2,658 cancer whole genomes
Erratum in
-
Author Correction: Analyses of non-coding somatic drivers in 2,658 cancer whole genomes.Nature. 2023 Feb;614(7948):E40. doi: 10.1038/s41586-022-05599-9. Nature. 2023. PMID: 36697832 Free PMC article. No abstract available.
Abstract
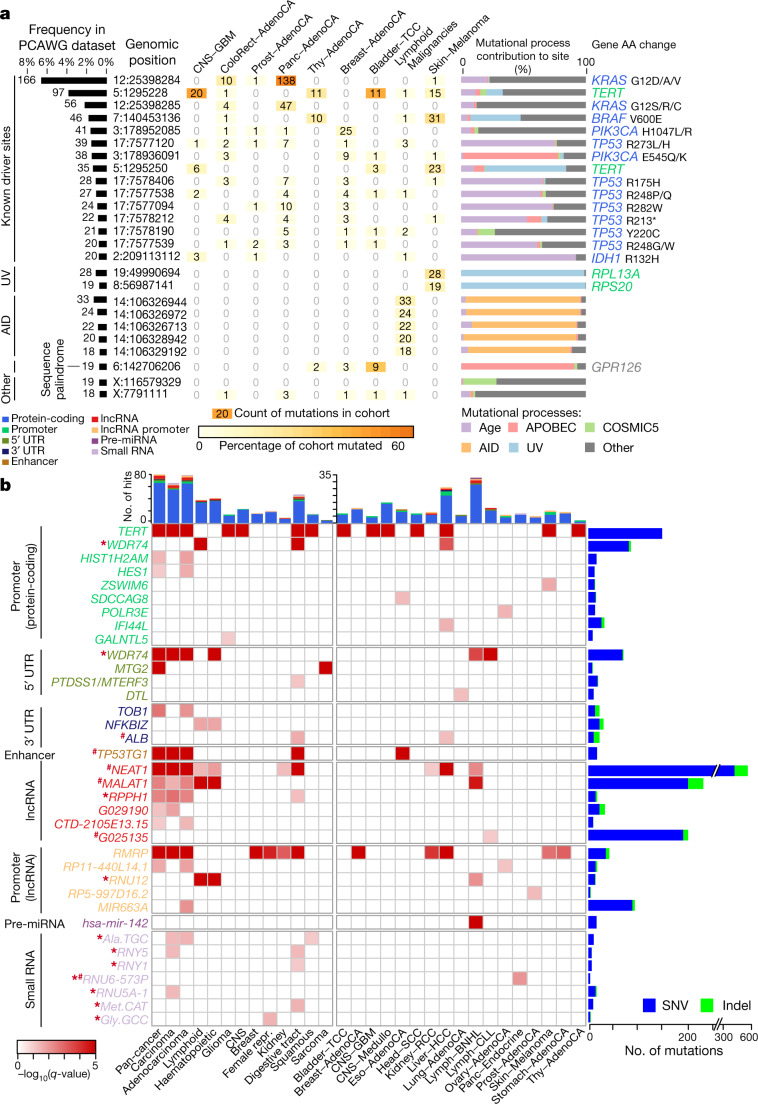
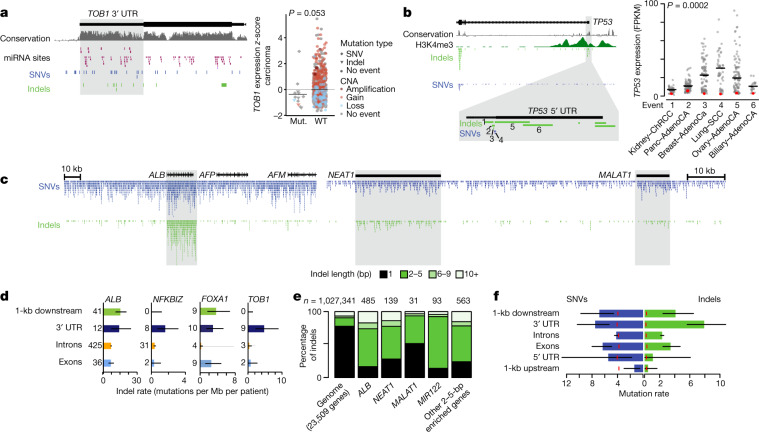
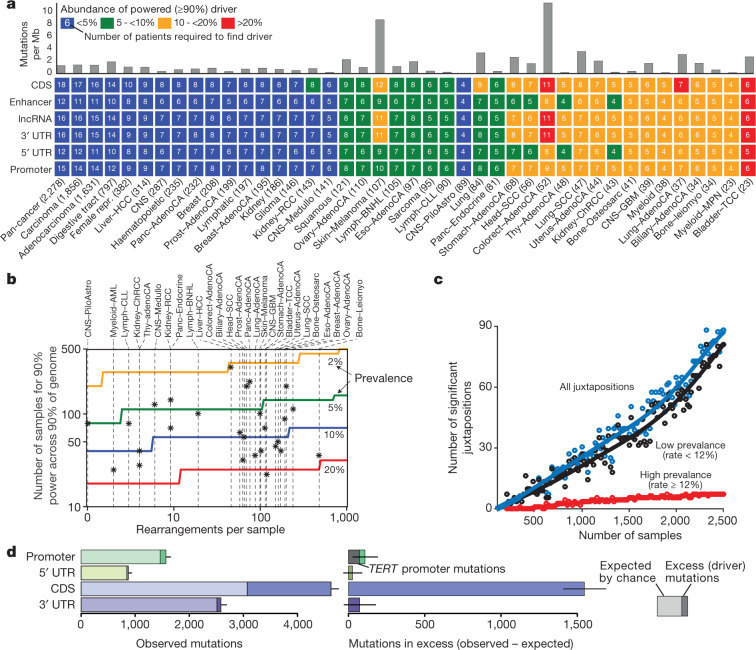
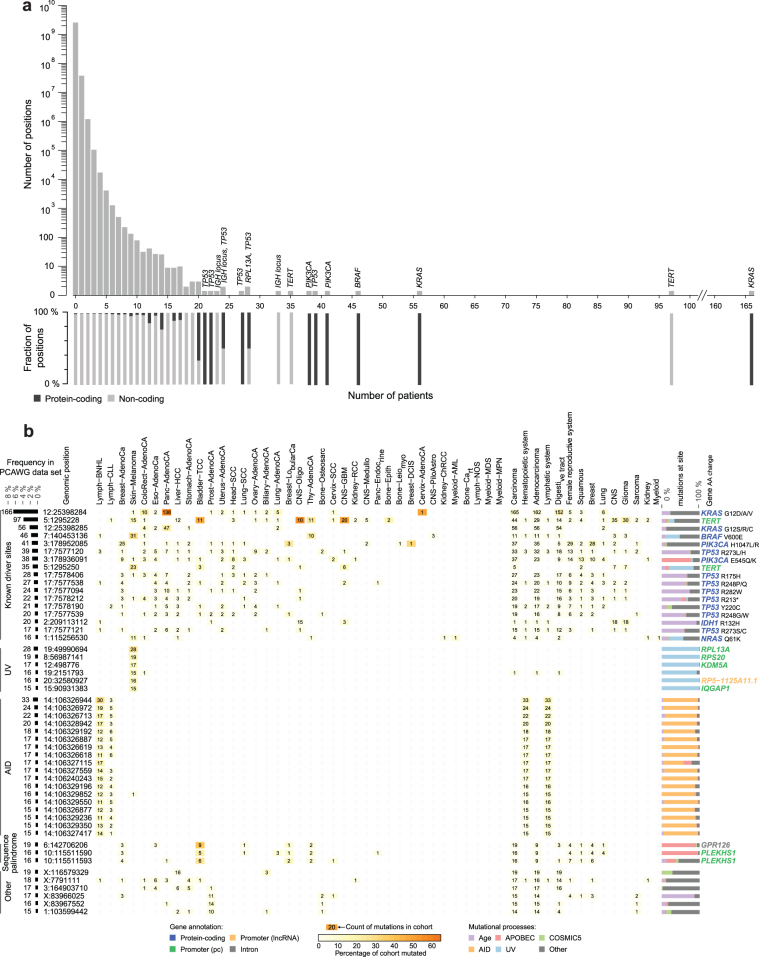
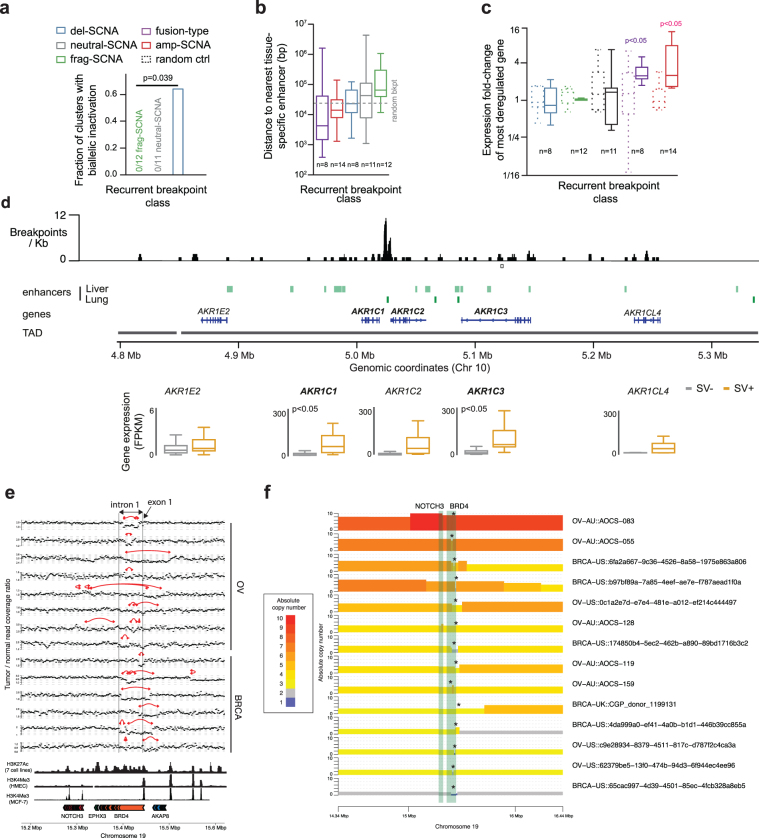
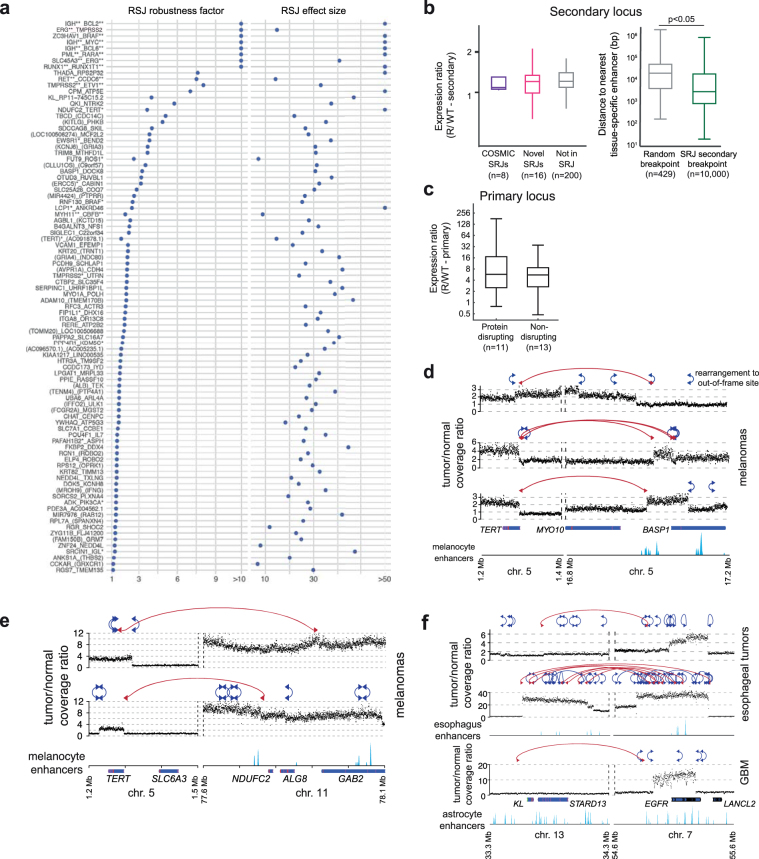
The discovery of drivers of cancer has traditionally focused on protein-coding genes1-4. Here we present analyses of driver point mutations and structural variants in non-coding regions across 2,658 genomes from the Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium5 of the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA). For point mutations, we developed a statistically rigorous strategy for combining significance levels from multiple methods of driver discovery that overcomes the limitations of individual methods. For structural variants, we present two methods of driver discovery, and identify regions that are significantly affected by recurrent breakpoints and recurrent somatic juxtapositions. Our analyses confirm previously reported drivers6,7, raise doubts about others and identify novel candidates, including point mutations in the 5' region of TP53, in the 3' untranslated regions of NFKBIZ and TOB1, focal deletions in BRD4 and rearrangements in the loci of AKR1C genes. We show that although point mutations and structural variants that drive cancer are less frequent in non-coding genes and regulatory sequences than in protein-coding genes, additional examples of these drivers will be found as more cancer genomes become available.
Conflict of interest statement
The following authors declare that they have competing interests. P.B. receives grant funding from Novartis from an unrelated project; R.B. owns equity in Ampressa Therapeutics and receives grant funding from Novartis; G.G. receives research funds from IBM and Pharmacyclics and is an inventor on patent applications related to MuTect, ABSOLUTE, MutSig, MSMuTect, MSMutSig and POLYSOLVER; B.J.R. is a consultant at and has ownership interest (including stock, patents and so on) in Medley Genomics; O.S. is currently an employee of Cedilla Therapeutics); and Y.L. is currently an employee of Seven Bridges Genomics.
Figures

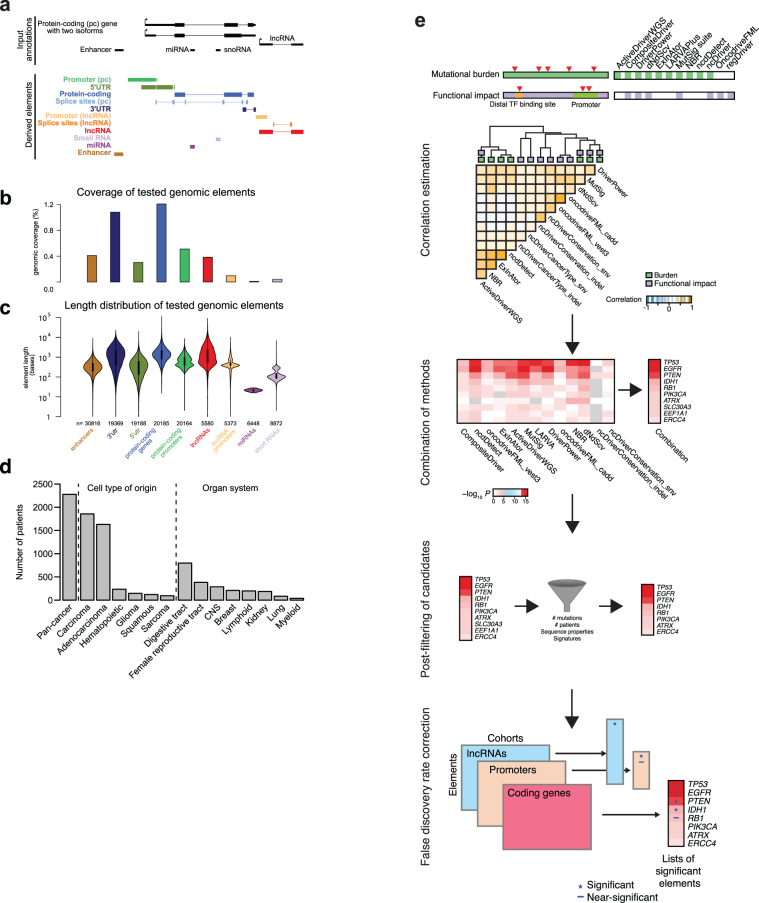
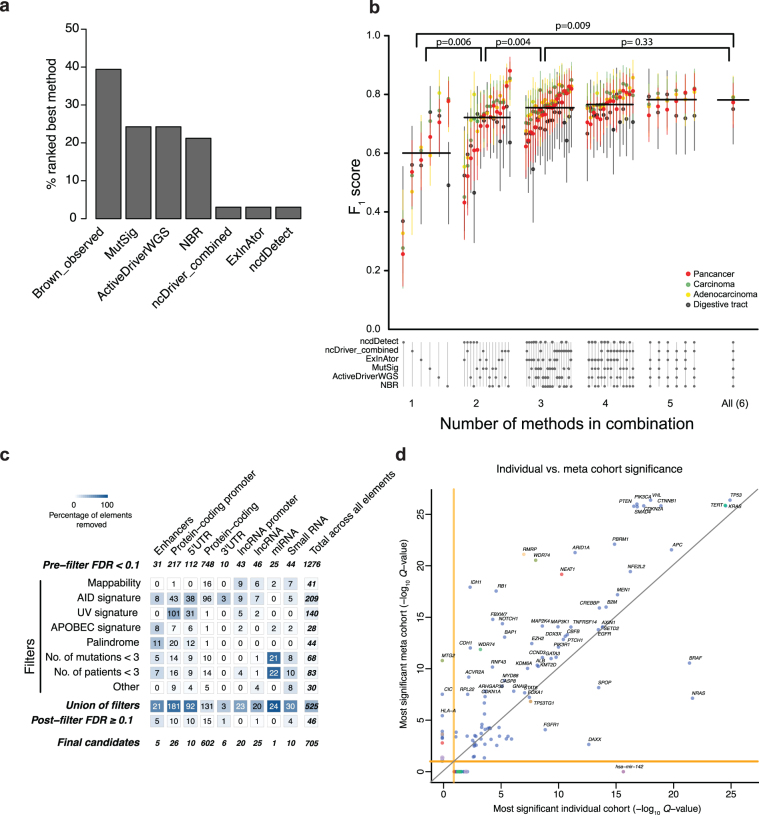
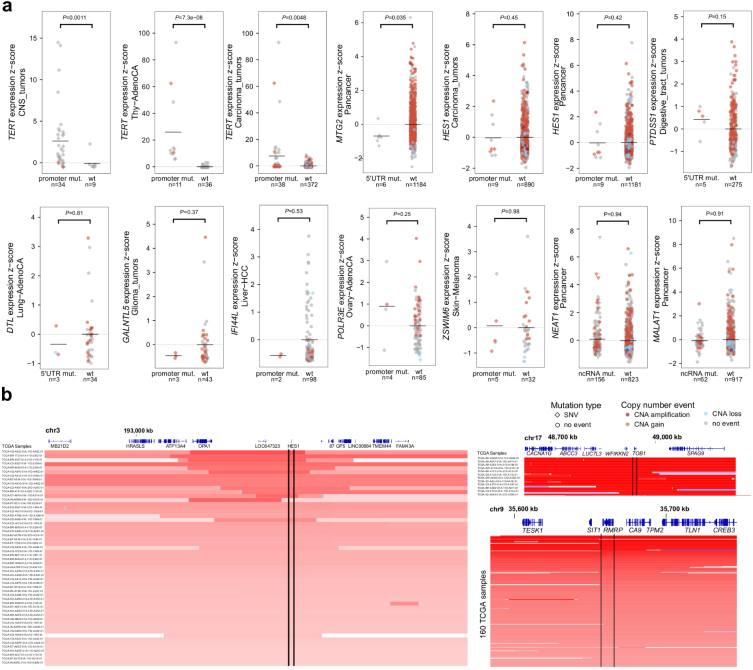

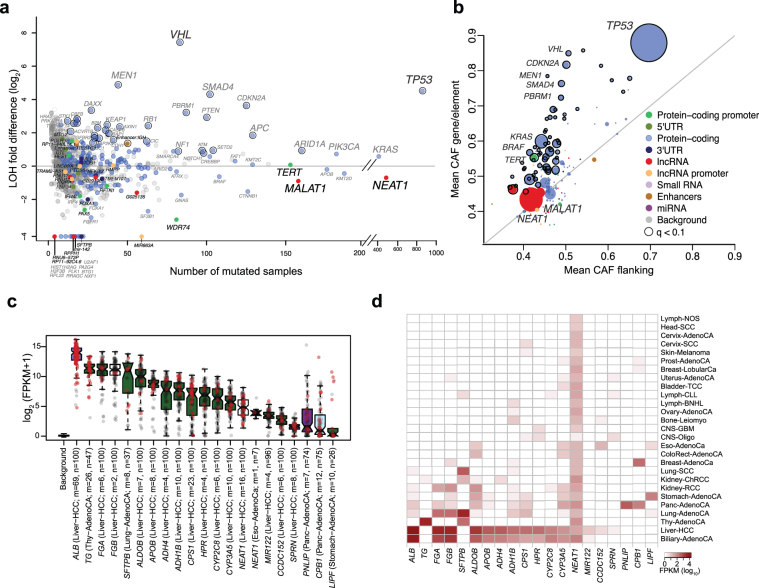



Comment in
-
Global genomics project unravels cancer's complexity at unprecedented scale.Nature. 2020 Feb;578(7793):39-40. doi: 10.1038/d41586-020-00213-2. Nature. 2020. PMID: 32025004 No abstract available.
Similar articles
-
Pan-cancer analysis of whole genomes.Nature. 2020 Feb;578(7793):82-93. doi: 10.1038/s41586-020-1969-6. Epub 2020 Feb 5. Nature. 2020. PMID: 32025007 Free PMC article.
-
Pathway and network analysis of more than 2500 whole cancer genomes.Nat Commun. 2020 Feb 5;11(1):729. doi: 10.1038/s41467-020-14367-0. Nat Commun. 2020. PMID: 32024854 Free PMC article.
-
Combined burden and functional impact tests for cancer driver discovery using DriverPower.Nat Commun. 2020 Feb 5;11(1):734. doi: 10.1038/s41467-019-13929-1. Nat Commun. 2020. PMID: 32024818 Free PMC article.
-
Decoding human cancer with whole genome sequencing: a review of PCAWG Project studies published in February 2020.Cancer Metastasis Rev. 2021 Sep;40(3):909-924. doi: 10.1007/s10555-021-09969-z. Epub 2021 Jun 7. Cancer Metastasis Rev. 2021. PMID: 34097189 Free PMC article. Review.
-
Beyond the exome: the role of non-coding somatic mutations in cancer.Ann Oncol. 2016 Feb;27(2):240-8. doi: 10.1093/annonc/mdv561. Epub 2015 Nov 23. Ann Oncol. 2016. PMID: 26598542 Review.
Cited by
-
Large-scale multi-omic analysis identifies noncoding somatic driver mutations and nominates ZFP36L2 as a driver gene for pancreatic ductal adenocarcinoma.medRxiv [Preprint]. 2024 Sep 24:2024.09.22.24314165. doi: 10.1101/2024.09.22.24314165. medRxiv. 2024. PMID: 39371173 Free PMC article. Preprint.
-
Worldwide analysis of actionable genomic alterations in lung cancer and targeted pharmacogenomic strategies.Heliyon. 2024 Sep 5;10(17):e37488. doi: 10.1016/j.heliyon.2024.e37488. eCollection 2024 Sep 15. Heliyon. 2024. PMID: 39296198 Free PMC article.
-
Integrative identification of non-coding regulatory regions driving metastatic prostate cancer.Cell Rep. 2024 Sep 24;43(9):114764. doi: 10.1016/j.celrep.2024.114764. Epub 2024 Sep 13. Cell Rep. 2024. PMID: 39276353 Free PMC article.
-
Long Non-Coding RNAs, Nuclear Receptors and Their Cross-Talks in Cancer-Implications and Perspectives.Cancers (Basel). 2024 Aug 22;16(16):2920. doi: 10.3390/cancers16162920. Cancers (Basel). 2024. PMID: 39199690 Free PMC article. Review.
-
Unveil Intrahepatic Cholangiocarcinoma Heterogeneity through the Lens of Omics and Multi-Omics Approaches.Cancers (Basel). 2024 Aug 20;16(16):2889. doi: 10.3390/cancers16162889. Cancers (Basel). 2024. PMID: 39199659 Free PMC article. Review.
References
-
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Network. Pan-cancer analysis of whole genomes. Nature 10.1038/s41586-020-1969-6 (2020).
Publication types
MeSH terms
Grants and funding
- R01 CA188228/CA/NCI NIH HHS/United States
- U54 CA143798/CA/NCI NIH HHS/United States
- R01 CA095175/CA/NCI NIH HHS/United States
- R01 HG007069/HG/NHGRI NIH HHS/United States
- T32 HG002295/HG/NHGRI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R01 CA218668/CA/NCI NIH HHS/United States
- T32 GM008313/GM/NIGMS NIH HHS/United States
- U24 CA210999/CA/NCI NIH HHS/United States
- R01 CA217991/CA/NCI NIH HHS/United States
- R01 CA219943/CA/NCI NIH HHS/United States
- U24 CA210990/CA/NCI NIH HHS/United States
- R35 GM127029/GM/NIGMS NIH HHS/United States
- U24 CA211000/CA/NCI NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- 21777/CRUK_/Cancer Research UK/United Kingdom
- U24 CA143845/CA/NCI NIH HHS/United States
- R01 CA215489/CA/NCI NIH HHS/United States
- T32 GM007205/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
