

How to choose templates for modeling of protein complexes: Insights from benchmarking template-based docking
- PMID: 31994759
- PMCID: PMC7375009
- DOI: 10.1002/prot.25875
How to choose templates for modeling of protein complexes: Insights from benchmarking template-based docking
Abstract
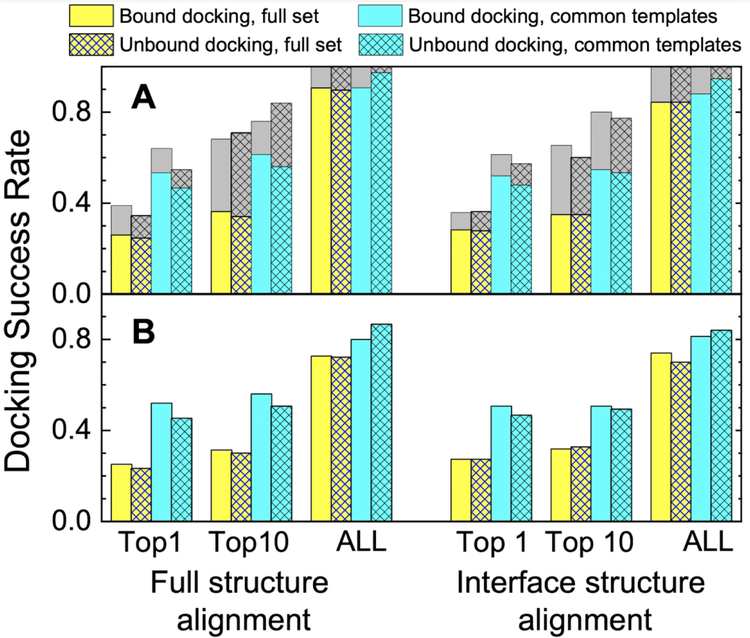
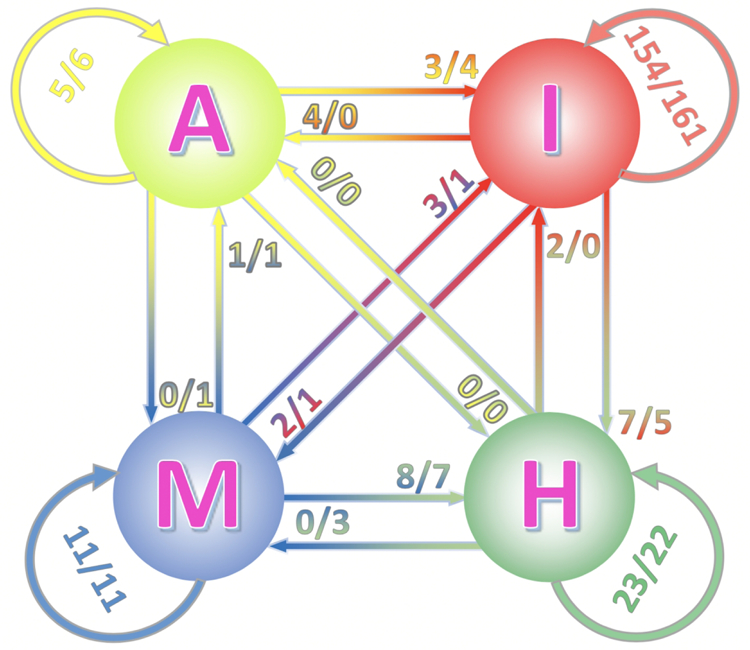
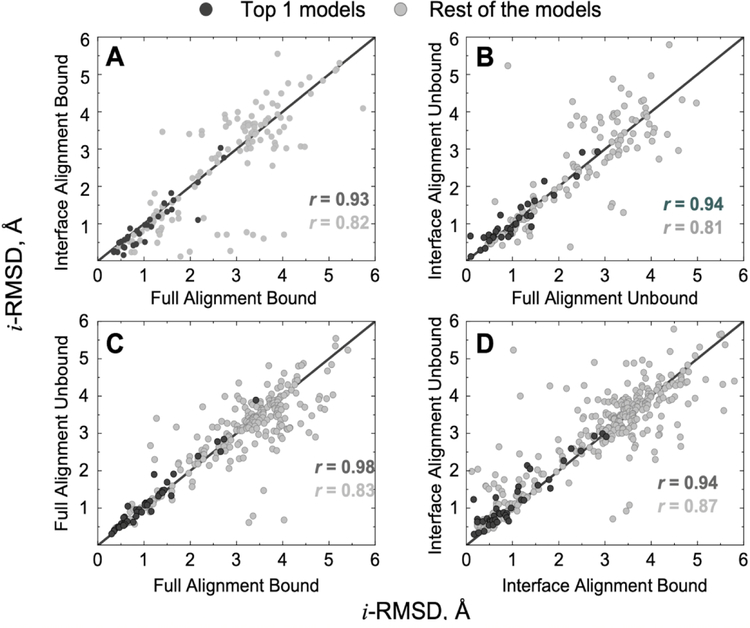
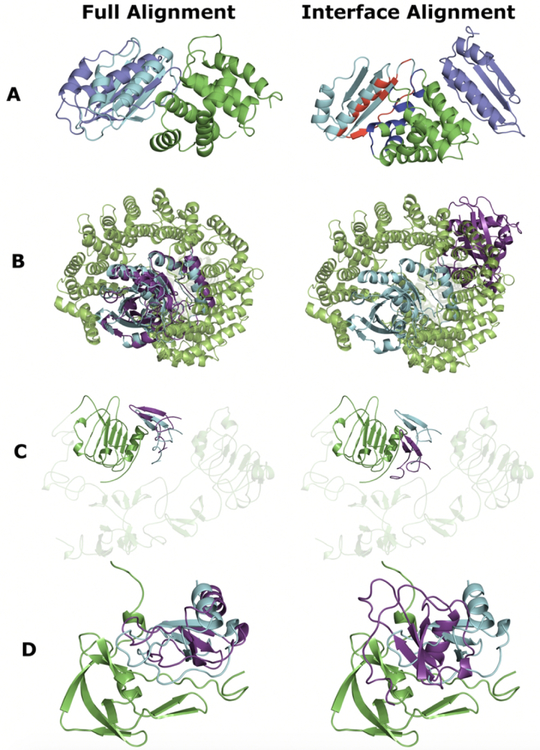
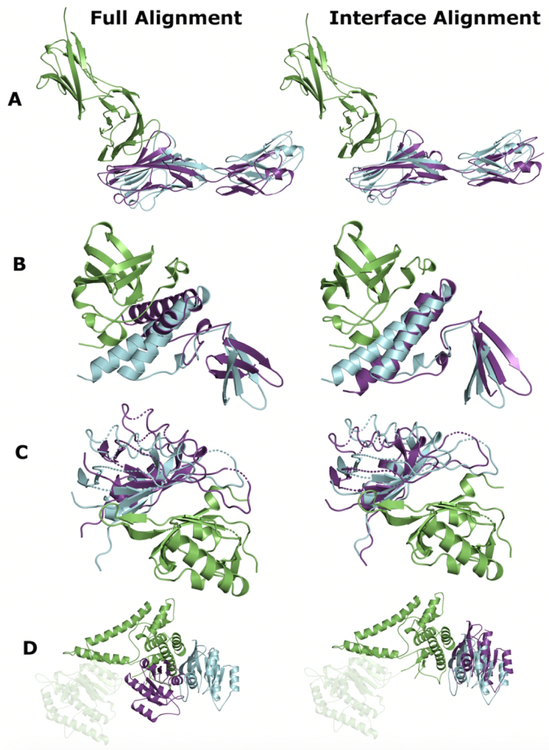
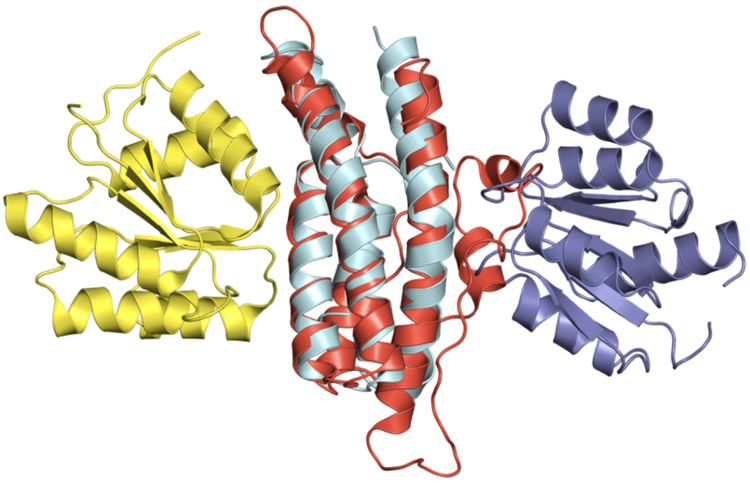
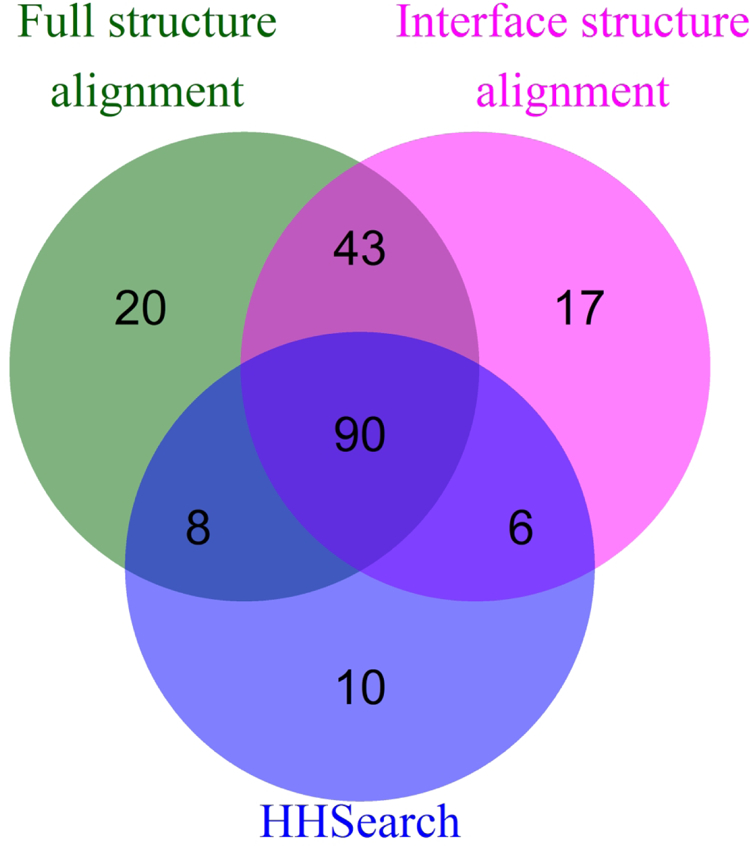
Comparative docking is based on experimentally determined structures of protein-protein complexes (templates), following the paradigm that proteins with similar sequences and/or structures form similar complexes. Modeling utilizing structure similarity of target monomers to template complexes significantly expands structural coverage of the interactome. Template-based docking by structure alignment can be performed for the entire structures or by aligning targets to the bound interfaces of the experimentally determined complexes. Systematic benchmarking of docking protocols based on full and interface structure alignment showed that both protocols perform similarly, with top 1 docking success rate 26%. However, in terms of the models' quality, the interface-based docking performed marginally better. The interface-based docking is preferable when one would suspect a significant conformational change in the full protein structure upon binding, for example, a rearrangement of the domains in multidomain proteins. Importantly, if the same structure is selected as the top template by both full and interface alignment, the docking success rate increases 2-fold for both top 1 and top 10 predictions. Matching structural annotations of the target and template proteins for template detection, as a computationally less expensive alternative to structural alignment, did not improve the docking performance. Sophisticated remote sequence homology detection added templates to the pool of those identified by structure-based alignment, suggesting that for practical docking, the combination of the structure alignment protocols and the remote sequence homology detection may be useful in order to avoid potential flaws in generation of the structural templates library.
Keywords: protein recognition; sequence homology; structure prediction; structure similarity; template detection.
© 2020 Wiley Periodicals, Inc.
Figures
Similar articles
-
ClusPro in rounds 38 to 45 of CAPRI: Toward combining template-based methods with free docking.Proteins. 2020 Aug;88(8):1082-1090. doi: 10.1002/prot.25887. Epub 2020 Mar 23. Proteins. 2020. PMID: 32142178 Free PMC article.
-
Structure prediction of biological assemblies using GALAXY in CAPRI rounds 38-45.Proteins. 2020 Aug;88(8):1009-1017. doi: 10.1002/prot.25859. Epub 2019 Dec 10. Proteins. 2020. PMID: 31774573
-
Docking proteins and peptides under evolutionary constraints in Critical Assessment of PRediction of Interactions rounds 38 to 45.Proteins. 2020 Aug;88(8):986-998. doi: 10.1002/prot.25857. Epub 2019 Dec 3. Proteins. 2020. PMID: 31746034
-
What method to use for protein-protein docking?Curr Opin Struct Biol. 2019 Apr;55:1-7. doi: 10.1016/j.sbi.2018.12.010. Epub 2019 Feb 1. Curr Opin Struct Biol. 2019. PMID: 30711743 Free PMC article. Review.
-
Advances in template-based protein docking by utilizing interfaces towards completing structural interactome.Curr Opin Struct Biol. 2015 Dec;35:87-92. doi: 10.1016/j.sbi.2015.10.001. Epub 2015 Nov 9. Curr Opin Struct Biol. 2015. PMID: 26539658 Review.
Cited by
-
AlphaFold predictions of fold-switched conformations are driven by structure memorization.Nat Commun. 2024 Aug 24;15(1):7296. doi: 10.1038/s41467-024-51801-z. Nat Commun. 2024. PMID: 39181864 Free PMC article.
-
Characterization of epitranscriptome reader proteins experimentally and in silico: Current knowledge and future perspectives beyond the YTH domain.Comput Struct Biotechnol J. 2023 Jun 30;21:3541-3556. doi: 10.1016/j.csbj.2023.06.018. eCollection 2023. Comput Struct Biotechnol J. 2023. PMID: 37501707 Free PMC article. Review.
-
Structural motifs in protein cores and at protein-protein interfaces are different.Protein Sci. 2021 Feb;30(2):381-390. doi: 10.1002/pro.3996. Epub 2020 Nov 20. Protein Sci. 2021. PMID: 33166001 Free PMC article.
-
AlphaFold2 has more to learn about protein energy landscapes.bioRxiv [Preprint]. 2023 Dec 13:2023.12.12.571380. doi: 10.1101/2023.12.12.571380. bioRxiv. 2023. Update in: Nat Commun. 2024 Aug 24;15(1):7296. doi: 10.1038/s41467-024-51801-z PMID: 38168383 Free PMC article. Updated. Preprint.
-
Challenges in protein docking.Curr Opin Struct Biol. 2020 Oct;64:160-165. doi: 10.1016/j.sbi.2020.07.001. Epub 2020 Aug 21. Curr Opin Struct Biol. 2020. PMID: 32836051 Free PMC article. Review.
References
-
- Aloy P, Ceulemans H, Stark A, Russell RB. The relationship between sequence and interaction divergence in proteins. J Mol Biol 2003;332:989–998. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
