

Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan
- PMID: 31987001
- PMCID: PMC7067204
- DOI: 10.1080/22221751.2020.1719902
Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan
Erratum in
-
Correction.Emerg Microbes Infect. 2020 Mar 5;9(1):540. doi: 10.1080/22221751.2020.1737364. eCollection 2020. Emerg Microbes Infect. 2020. PMID: 32133926 Free PMC article. No abstract available.
Abstract
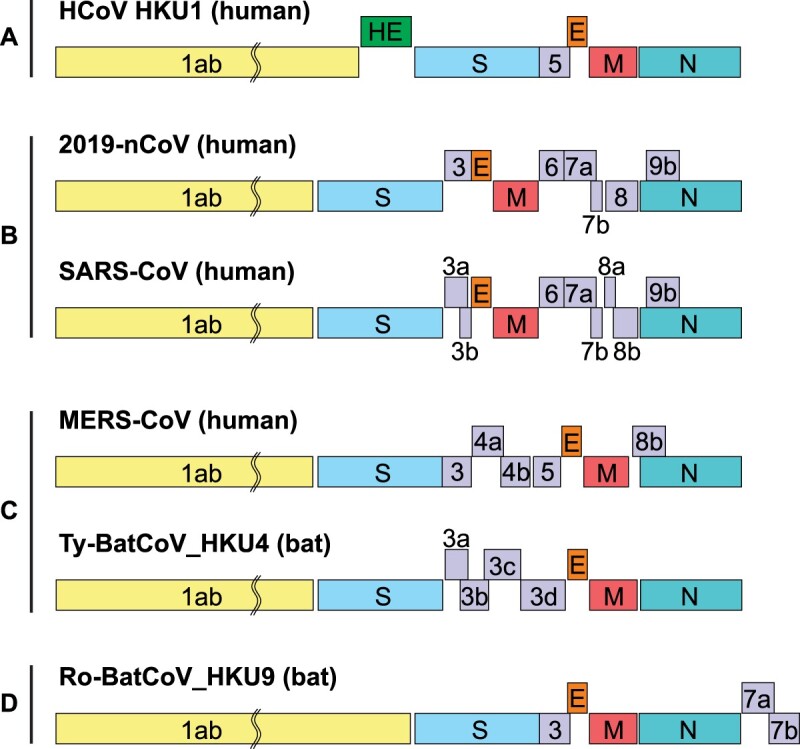
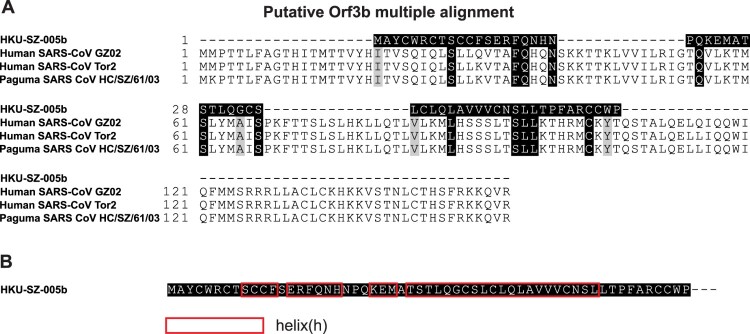
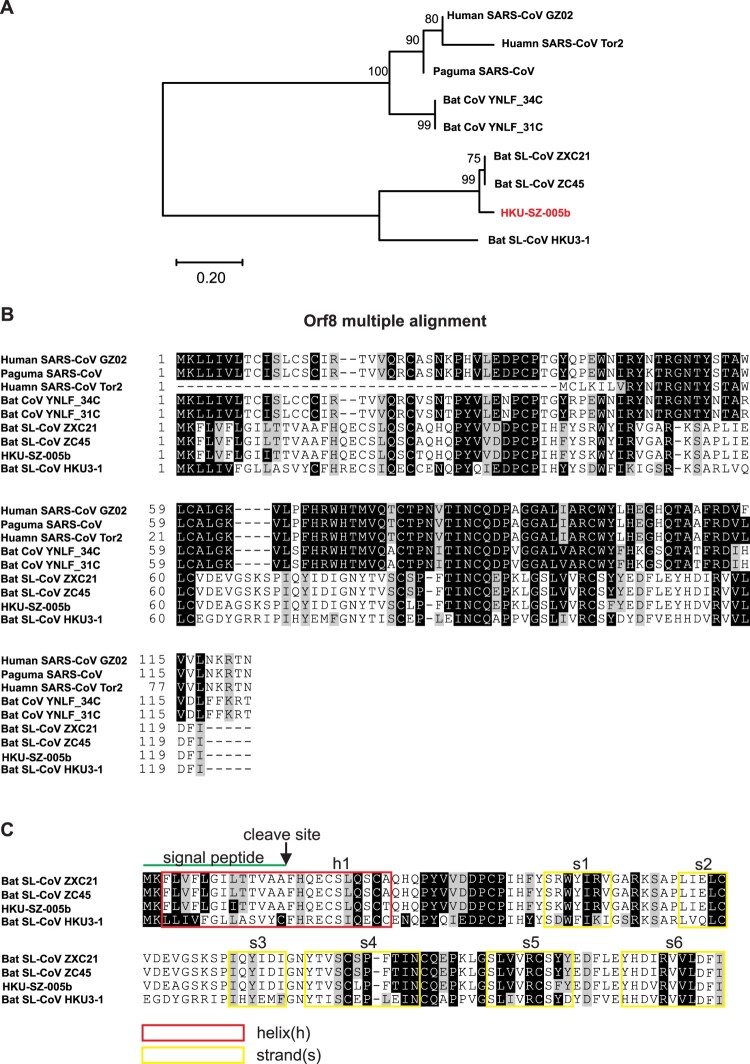
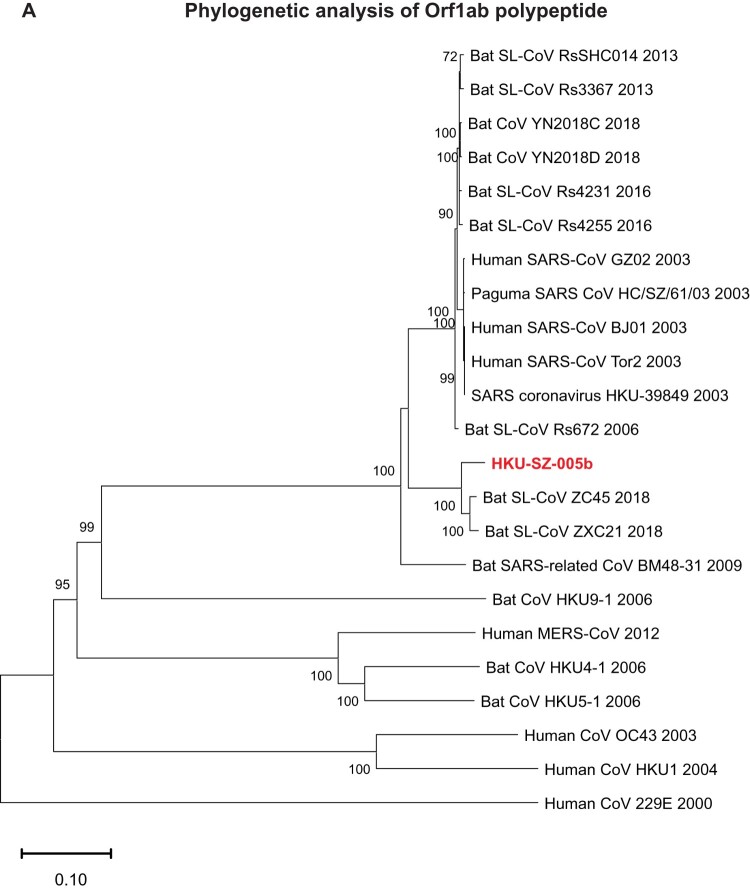
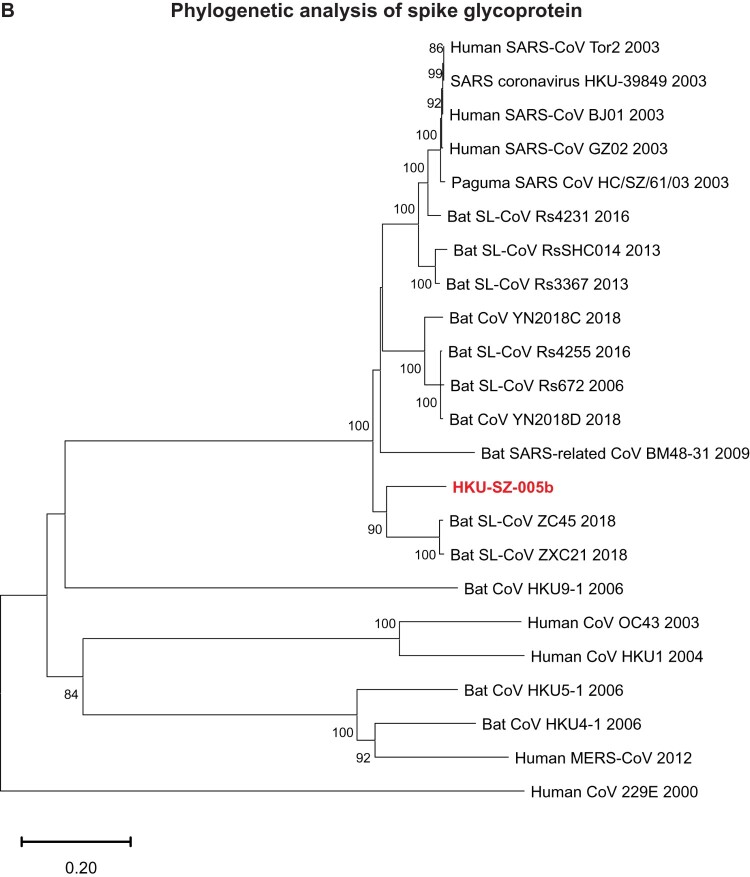
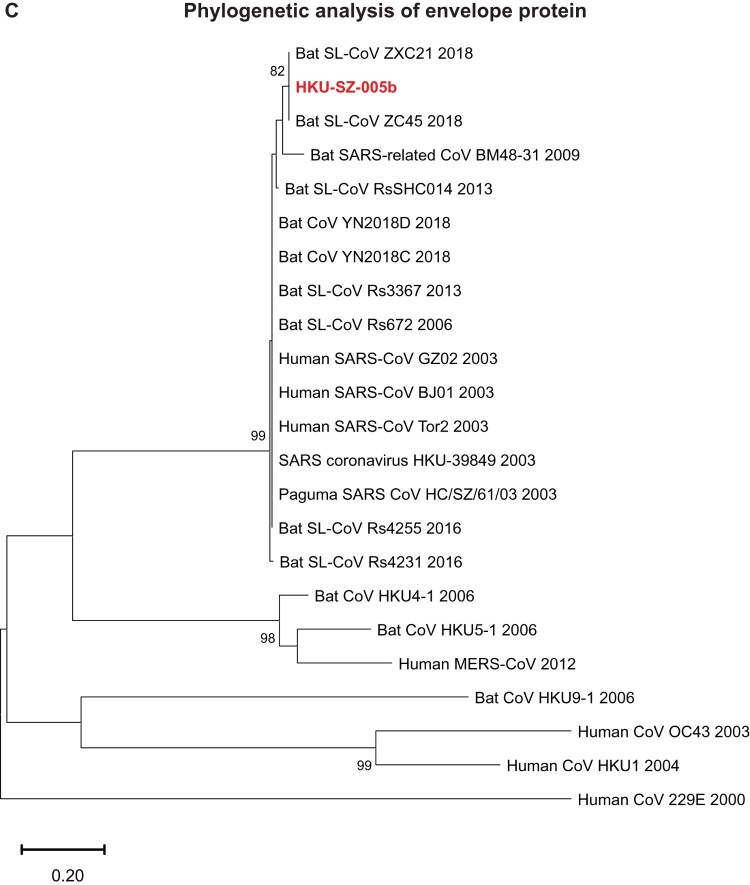
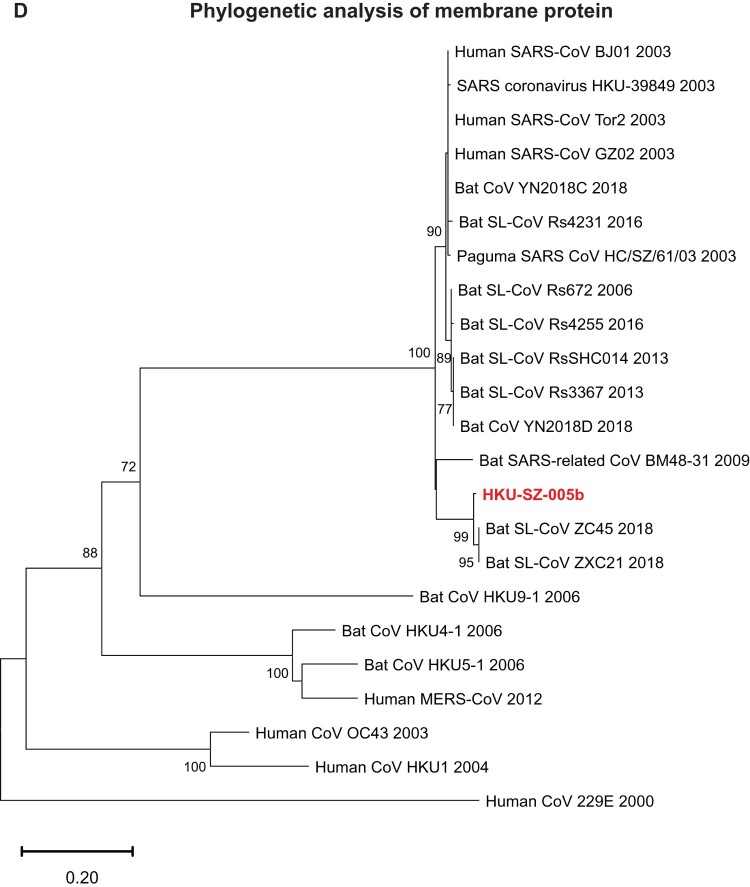
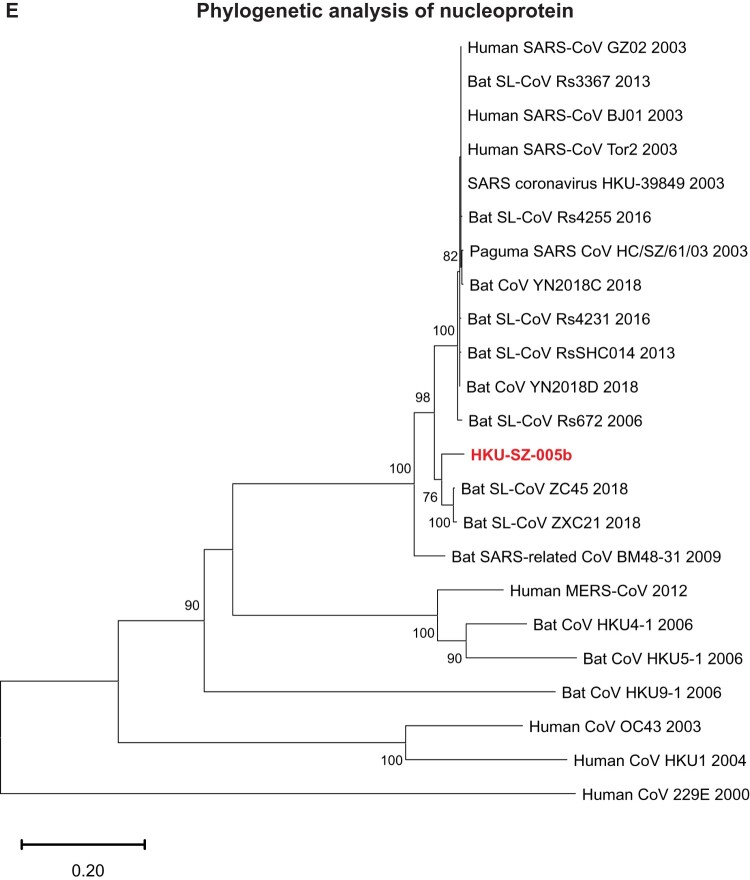
A mysterious outbreak of atypical pneumonia in late 2019 was traced to a seafood wholesale market in Wuhan of China. Within a few weeks, a novel coronavirus tentatively named as 2019 novel coronavirus (2019-nCoV) was announced by the World Health Organization. We performed bioinformatics analysis on a virus genome from a patient with 2019-nCoV infection and compared it with other related coronavirus genomes. Overall, the genome of 2019-nCoV has 89% nucleotide identity with bat SARS-like-CoVZXC21 and 82% with that of human SARS-CoV. The phylogenetic trees of their orf1a/b, Spike, Envelope, Membrane and Nucleoprotein also clustered closely with those of the bat, civet and human SARS coronaviruses. However, the external subdomain of Spike's receptor binding domain of 2019-nCoV shares only 40% amino acid identity with other SARS-related coronaviruses. Remarkably, its orf3b encodes a completely novel short protein. Furthermore, its new orf8 likely encodes a secreted protein with an alpha-helix, following with a beta-sheet(s) containing six strands. Learning from the roles of civet in SARS and camel in MERS, hunting for the animal source of 2019-nCoV and its more ancestral virus would be important for understanding the origin and evolution of this novel lineage B betacoronavirus. These findings provide the basis for starting further studies on the pathogenesis, and optimizing the design of diagnostic, antiviral and vaccination strategies for this emerging infection.
Keywords: Coronavirus; SARS; Wuhan; bioinformatics; emerging; genome; respiratory; virus.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures




Similar articles
-
Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding.Lancet. 2020 Feb 22;395(10224):565-574. doi: 10.1016/S0140-6736(20)30251-8. Epub 2020 Jan 30. Lancet. 2020. PMID: 32007145 Free PMC article.
-
2019-nCoV (Wuhan virus), a novel Coronavirus: human-to-human transmission, travel-related cases, and vaccine readiness.J Infect Dev Ctries. 2020 Jan 31;14(1):3-17. doi: 10.3855/jidc.12425. J Infect Dev Ctries. 2020. PMID: 32088679
-
RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak.Emerg Microbes Infect. 2020 Feb 5;9(1):313-319. doi: 10.1080/22221751.2020.1725399. eCollection 2020. Emerg Microbes Infect. 2020. PMID: 32020836 Free PMC article.
-
Properties of Coronavirus and SARS-CoV-2.Malays J Pathol. 2020 Apr;42(1):3-11. Malays J Pathol. 2020. PMID: 32342926 Review.
-
[Etiology of epidemic outbreaks COVID-19 on Wuhan, Hubei province, Chinese People Republic associated with 2019-nCoV (Nidovirales, Coronaviridae, Coronavirinae, Betacoronavirus, Subgenus Sarbecovirus): lessons of SARS-CoV outbreak.].Vopr Virusol. 2020;65(1):6-15. doi: 10.36233/0507-4088-2020-65-1-6-15. Vopr Virusol. 2020. PMID: 32496715 Review. Russian.
Cited by
-
Electron Tomography as a Tool to Study SARS-CoV-2 Morphology.Int J Mol Sci. 2024 Nov 1;25(21):11762. doi: 10.3390/ijms252111762. Int J Mol Sci. 2024. PMID: 39519314 Free PMC article. Review.
-
Analysis of the structure and interactions of the SARS-CoV-2 ORF7b accessory protein.Proc Natl Acad Sci U S A. 2024 Nov 12;121(46):e2407731121. doi: 10.1073/pnas.2407731121. Epub 2024 Nov 7. Proc Natl Acad Sci U S A. 2024. PMID: 39508769 Free PMC article.
-
Current Updates on Variants of SARS-CoV- 2: Systematic Review.Health Sci Rep. 2024 Nov 4;7(11):e70166. doi: 10.1002/hsr2.70166. eCollection 2024 Nov. Health Sci Rep. 2024. PMID: 39502131 Free PMC article. Review.
-
A CRISPR-Cas13b System Degrades SARS-CoV and SARS-CoV-2 RNA In Vitro.Viruses. 2024 Sep 28;16(10):1539. doi: 10.3390/v16101539. Viruses. 2024. PMID: 39459873 Free PMC article.
-
Obesity as a Risk Factor for the Severity of COVID-19 in Pediatric Patients: Possible Mechanisms-A Narrative Review.Children (Basel). 2024 Sep 30;11(10):1203. doi: 10.3390/children11101203. Children (Basel). 2024. PMID: 39457167 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous