

Prion protein glycans reduce intracerebral fibril formation and spongiosis in prion disease
- PMID: 31985492
- PMCID: PMC7269597
- DOI: 10.1172/JCI131564
Prion protein glycans reduce intracerebral fibril formation and spongiosis in prion disease
Abstract
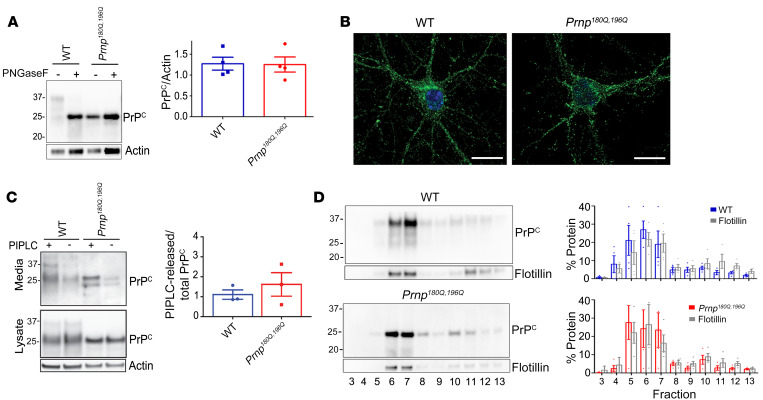
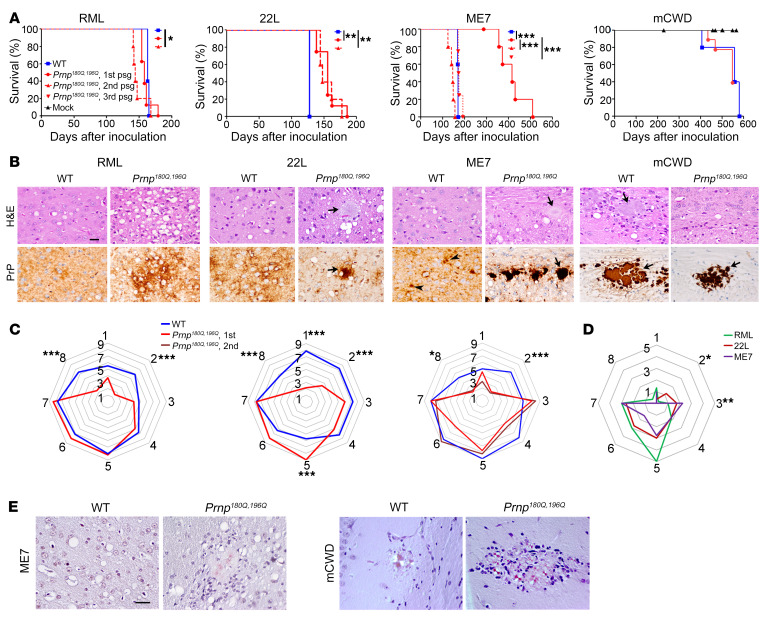
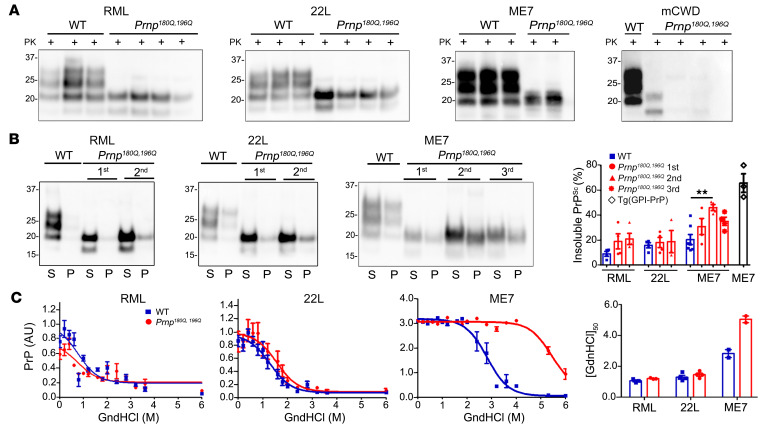
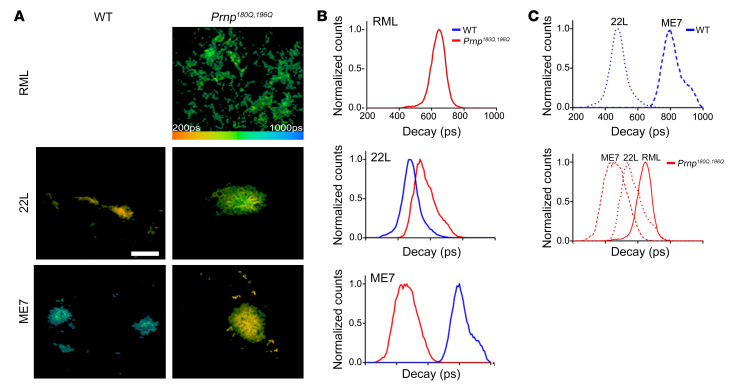
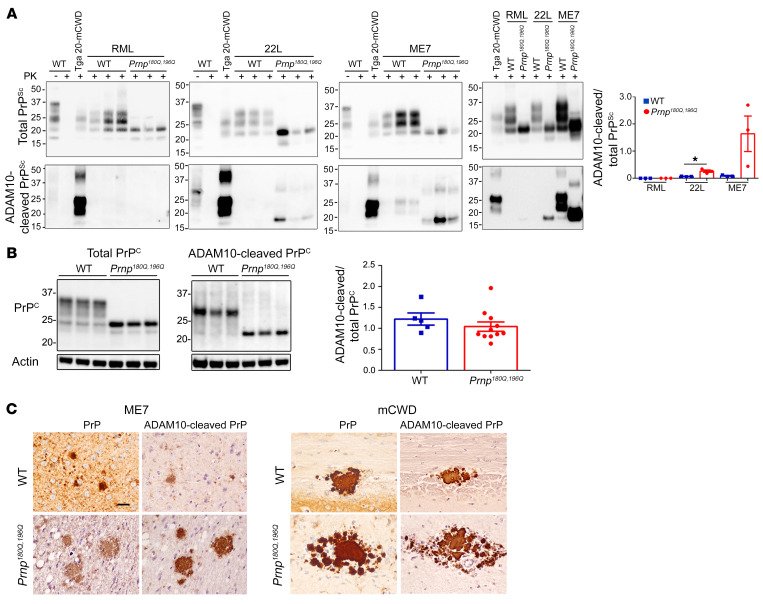
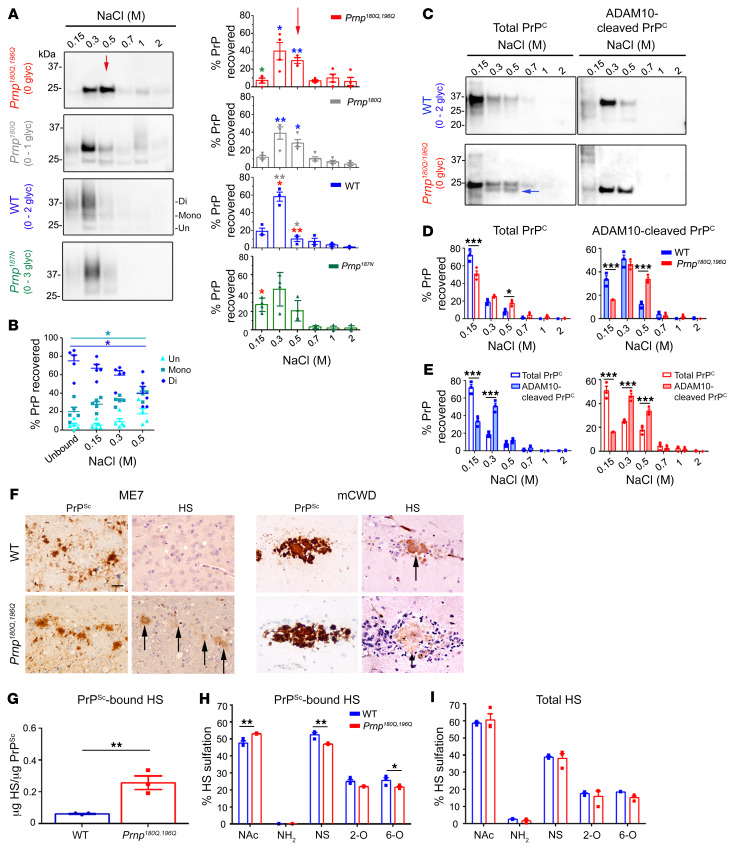
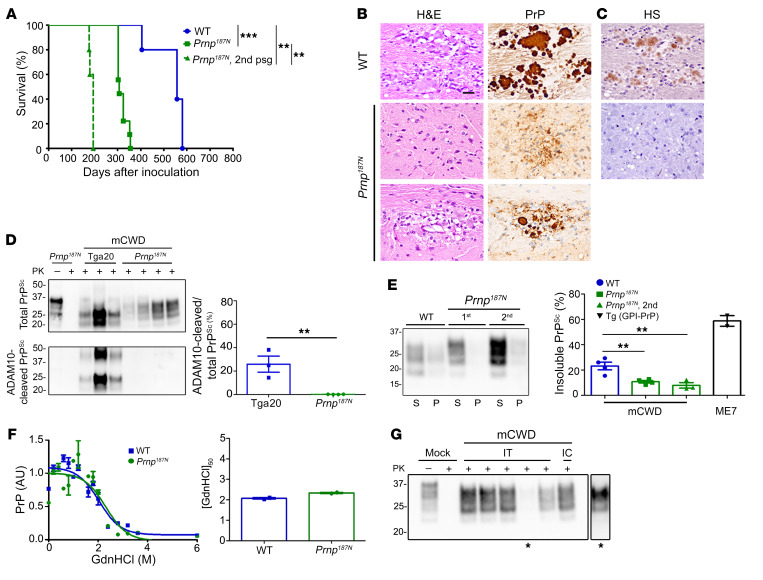
Posttranslational modifications (PTMs) are common among proteins that aggregate in neurodegenerative disease, yet how PTMs impact the aggregate conformation and disease progression remains unclear. By engineering knockin mice expressing prion protein (PrP) lacking 2 N-linked glycans (Prnp180Q/196Q), we provide evidence that glycans reduce spongiform degeneration and hinder plaque formation in prion disease. Prnp180Q/196Q mice challenged with 2 subfibrillar, non-plaque-forming prion strains instead developed plaques highly enriched in ADAM10-cleaved PrP and heparan sulfate (HS). Intriguingly, a third strain composed of intact, glycophosphatidylinositol-anchored (GPI-anchored) PrP was relatively unchanged, forming diffuse, HS-deficient deposits in both the Prnp180Q/196Q and WT mice, underscoring the pivotal role of the GPI-anchor in driving the aggregate conformation and disease phenotype. Finally, knockin mice expressing triglycosylated PrP (Prnp187N) challenged with a plaque-forming prion strain showed a phenotype reversal, with a striking disease acceleration and switch from plaques to predominantly diffuse, subfibrillar deposits. Our findings suggest that the dominance of subfibrillar aggregates in prion disease is due to the replication of GPI-anchored prions, with fibrillar plaques forming from poorly glycosylated, GPI-anchorless prions that interact with extracellular HS. These studies provide insight into how PTMs impact PrP interactions with polyanionic cofactors, and highlight PTMs as a major force driving the prion disease phenotype.
Keywords: Glycobiology; Infectious disease; Neurodegeneration; Neuroscience; Prions.
Conflict of interest statement
Figures
Comment in
-
Underglycosylated prion protein modulates plaque formation in the brain.J Clin Invest. 2020 Mar 2;130(3):1087-1089. doi: 10.1172/JCI134842. J Clin Invest. 2020. PMID: 31985491 Free PMC article.
Similar articles
-
Structural biology of ex vivo mammalian prions.J Biol Chem. 2022 Aug;298(8):102181. doi: 10.1016/j.jbc.2022.102181. Epub 2022 Jun 23. J Biol Chem. 2022. PMID: 35752366 Free PMC article. Review.
-
Prion protein post-translational modifications modulate heparan sulfate binding and limit aggregate size in prion disease.Neurobiol Dis. 2020 Aug;142:104955. doi: 10.1016/j.nbd.2020.104955. Epub 2020 May 24. Neurobiol Dis. 2020. PMID: 32454127 Free PMC article.
-
Shortening heparan sulfate chains prolongs survival and reduces parenchymal plaques in prion disease caused by mobile, ADAM10-cleaved prions.Acta Neuropathol. 2020 Mar;139(3):527-546. doi: 10.1007/s00401-019-02085-x. Epub 2019 Oct 31. Acta Neuropathol. 2020. PMID: 31673874 Free PMC article.
-
Enhanced neuroinvasion by smaller, soluble prions.Acta Neuropathol Commun. 2017 Apr 21;5(1):32. doi: 10.1186/s40478-017-0430-z. Acta Neuropathol Commun. 2017. PMID: 28431576 Free PMC article.
-
Prion protein-Semisynthetic prion protein (PrP) variants with posttranslational modifications.J Pept Sci. 2019 Oct;25(10):e3216. doi: 10.1002/psc.3216. J Pept Sci. 2019. PMID: 31713950 Free PMC article. Review.
Cited by
-
Efficient transmission of human prion diseases to a glycan-free prion protein-expressing host.Brain. 2024 Apr 4;147(4):1539-1552. doi: 10.1093/brain/awad399. Brain. 2024. PMID: 38000783 Free PMC article.
-
Cleavage site-directed antibodies reveal the prion protein in humans is shed by ADAM10 at Y226 and associates with misfolded protein deposits in neurodegenerative diseases.Acta Neuropathol. 2024 Jul 9;148(1):2. doi: 10.1007/s00401-024-02763-5. Acta Neuropathol. 2024. PMID: 38980441 Free PMC article.
-
N-glycans show distinct spatial distribution in mouse brain.Glycobiology. 2023 Dec 25;33(11):935-942. doi: 10.1093/glycob/cwad077. Glycobiology. 2023. PMID: 37792804 Free PMC article.
-
A novel subtype of sporadic Creutzfeldt-Jakob disease with PRNP codon 129MM genotype and PrP plaques.Acta Neuropathol. 2023 Jul;146(1):121-143. doi: 10.1007/s00401-023-02581-1. Epub 2023 May 8. Acta Neuropathol. 2023. PMID: 37156880 Free PMC article.
-
Structural biology of ex vivo mammalian prions.J Biol Chem. 2022 Aug;298(8):102181. doi: 10.1016/j.jbc.2022.102181. Epub 2022 Jun 23. J Biol Chem. 2022. PMID: 35752366 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
