

Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy
- PMID: 31984784
- PMCID: PMC7099488
- DOI: 10.1152/ajpgi.00223.2019
Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy
Abstract
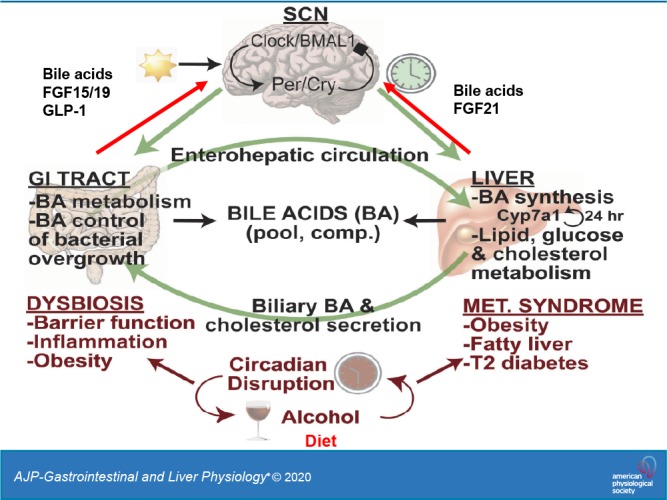
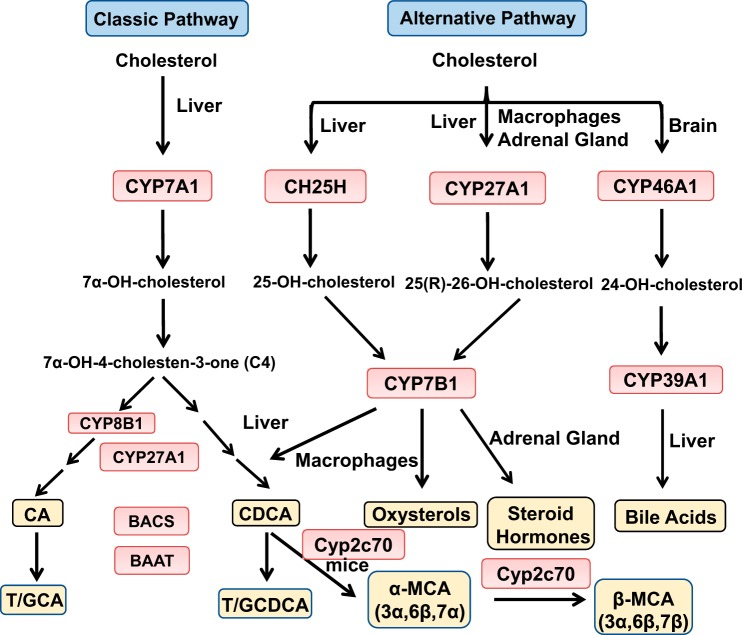
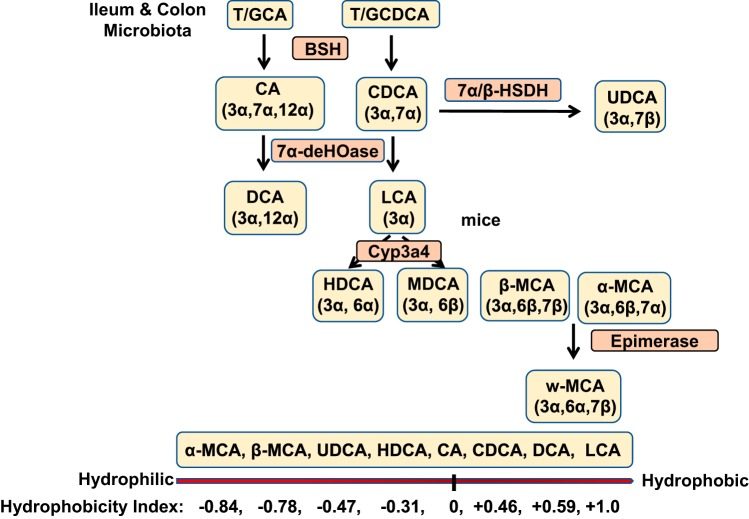
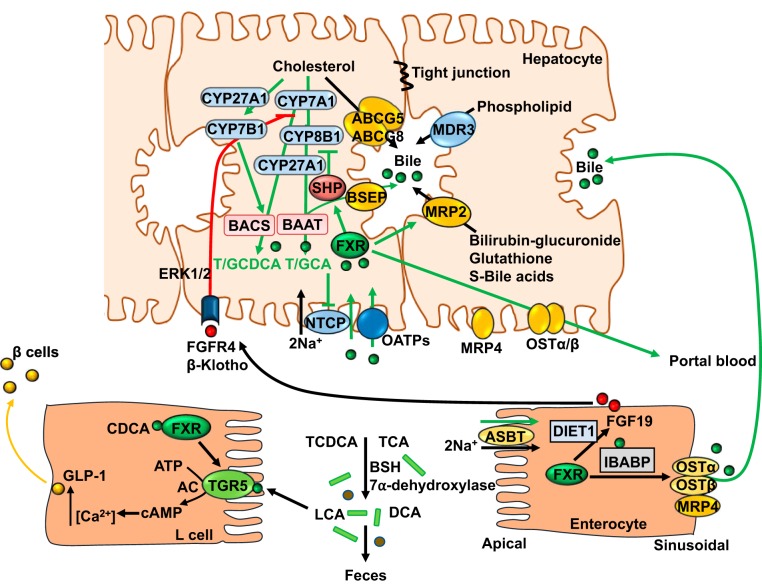
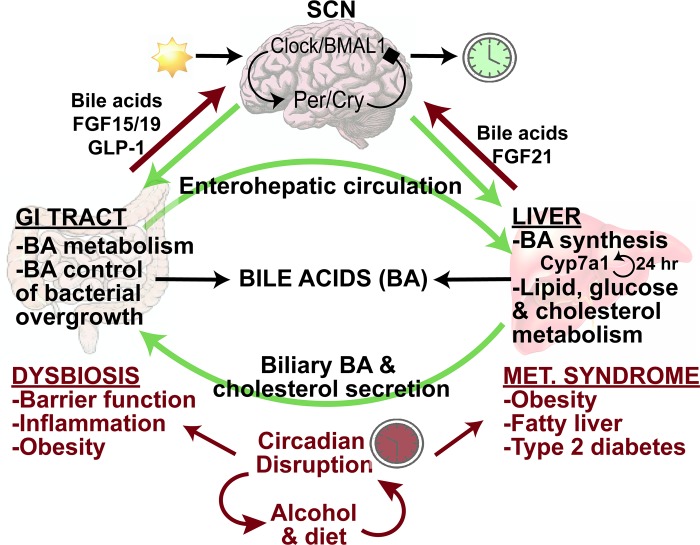
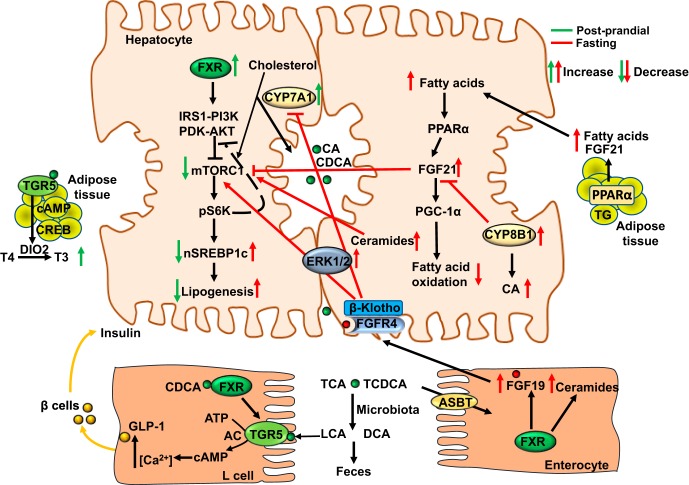
Bile acid synthesis is the most significant pathway for catabolism of cholesterol and for maintenance of whole body cholesterol homeostasis. Bile acids are physiological detergents that absorb, distribute, metabolize, and excrete nutrients, drugs, and xenobiotics. Bile acids also are signal molecules and metabolic integrators that activate nuclear farnesoid X receptor (FXR) and membrane Takeda G protein-coupled receptor 5 (TGR5; i.e., G protein-coupled bile acid receptor 1) to regulate glucose, lipid, and energy metabolism. The gut-to-liver axis plays a critical role in the transformation of primary bile acids to secondary bile acids, in the regulation of bile acid synthesis to maintain composition within the bile acid pool, and in the regulation of metabolic homeostasis to prevent hyperglycemia, dyslipidemia, obesity, and diabetes. High-fat and high-calorie diets, dysbiosis, alcohol, drugs, and disruption of sleep and circadian rhythms cause metabolic diseases, including alcoholic and nonalcoholic fatty liver diseases, obesity, diabetes, and cardiovascular disease. Bile acid-based drugs that target bile acid receptors are being developed for the treatment of metabolic diseases of the liver.
Keywords: Takeda G protein-coupled receptor 5; alcoholic and nonalcoholic fatty; bile acid metabolism; bile acid therapies; farnesoid X receptor; liver diseases.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
Similar articles
-
Farnesoid X receptor induces Takeda G-protein receptor 5 cross-talk to regulate bile acid synthesis and hepatic metabolism.J Biol Chem. 2017 Jun 30;292(26):11055-11069. doi: 10.1074/jbc.M117.784322. Epub 2017 May 6. J Biol Chem. 2017. PMID: 28478385 Free PMC article.
-
Bile acid receptors in non-alcoholic fatty liver disease.Biochem Pharmacol. 2013 Dec 1;86(11):1517-24. doi: 10.1016/j.bcp.2013.08.015. Epub 2013 Aug 26. Biochem Pharmacol. 2013. PMID: 23988487 Free PMC article. Review.
-
Intestinal Farnesoid X Receptor and Takeda G Protein Couple Receptor 5 Signaling in Metabolic Regulation.Dig Dis. 2017;35(3):241-245. doi: 10.1159/000450981. Epub 2017 Mar 1. Dig Dis. 2017. PMID: 28249273 Free PMC article. Review.
-
G-protein-coupled bile acid receptor plays a key role in bile acid metabolism and fasting-induced hepatic steatosis in mice.Hepatology. 2017 Mar;65(3):813-827. doi: 10.1002/hep.28707. Epub 2016 Jul 30. Hepatology. 2017. PMID: 27351453 Free PMC article.
-
Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease.Gastroenterology. 2017 May;152(7):1679-1694.e3. doi: 10.1053/j.gastro.2017.01.055. Epub 2017 Feb 15. Gastroenterology. 2017. PMID: 28214524 Review.
Cited by
-
Targeting Farnesoid X Receptor in Tumor and the Tumor Microenvironment: Implication for Therapy.Int J Mol Sci. 2023 Dec 19;25(1):6. doi: 10.3390/ijms25010006. Int J Mol Sci. 2023. PMID: 38203175 Free PMC article. Review.
-
Alismatis Rhizoma methanolic extract-Effects on metabolic syndrome and mechanisms of triterpenoids using a metabolomic and lipidomic approach.Front Pharmacol. 2022 Sep 9;13:983428. doi: 10.3389/fphar.2022.983428. eCollection 2022. Front Pharmacol. 2022. PMID: 36160458 Free PMC article.
-
Gut Microbiota Metabolites in NAFLD Pathogenesis and Therapeutic Implications.Int J Mol Sci. 2020 Jul 23;21(15):5214. doi: 10.3390/ijms21155214. Int J Mol Sci. 2020. PMID: 32717871 Free PMC article. Review.
-
NLRP3 Inflammasome and Gut Dysbiosis Linking Diabetes Mellitus and Inflammatory Bowel Disease.Arch Intern Med Res. 2024;7(3):200-218. doi: 10.26502/aimr.0178. Epub 2024 Aug 31. Arch Intern Med Res. 2024. PMID: 39328924 Free PMC article.
-
Integrated traditional Chinese and Western medicine in the prevention and treatment of non-alcoholic fatty liver disease: future directions and strategies.Chin Med. 2024 Feb 3;19(1):21. doi: 10.1186/s13020-024-00894-1. Chin Med. 2024. PMID: 38310315 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources