

Herpesviruses and the Unfolded Protein Response
- PMID: 31877732
- PMCID: PMC7019427
- DOI: 10.3390/v12010017
Herpesviruses and the Unfolded Protein Response
Abstract
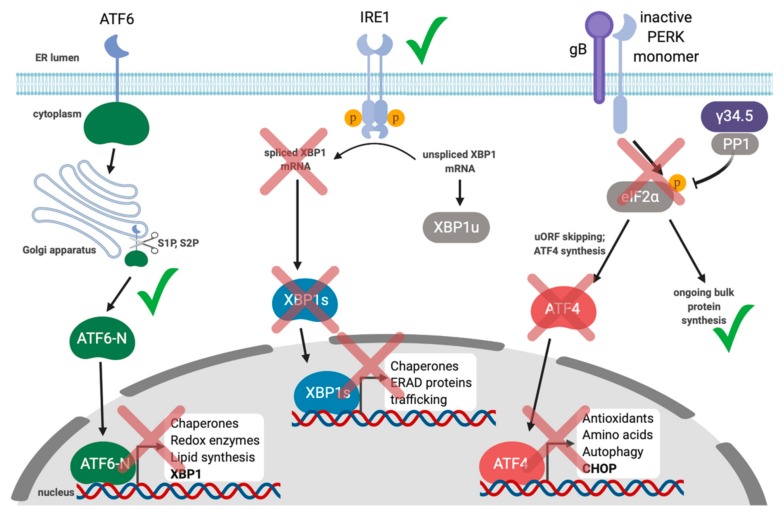
Herpesviruses usurp cellular stress responses to promote viral replication and avoid immune surveillance. The unfolded protein response (UPR) is a conserved stress response that is activated when the protein load in the ER exceeds folding capacity and misfolded proteins accumulate. The UPR aims to restore protein homeostasis through translational and transcriptional reprogramming; if homeostasis cannot be restored, the UPR switches from "helper" to "executioner", triggering apoptosis. It is thought that the burst of herpesvirus glycoprotein synthesis during lytic replication causes ER stress, and that these viruses may have evolved mechanisms to manage UPR signaling to create an optimal niche for replication. The past decade has seen considerable progress in understanding how herpesviruses reprogram the UPR. Here we provide an overview of the molecular events of UPR activation, signaling and transcriptional outputs, and highlight key evidence that herpesviruses hijack the UPR to aid infection.
Keywords: ATF4; ATF6; GADD34; IRE1; Kaposi’s sarcoma-associated herpesvirus (KSHV); PERK; XBP1; cytomegalovirus (CMV); herpes simplex virus (HSV); herpesvirus; integrated stress response (ISR); unfolded protein response (UPR).
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the writing of the manuscript.
Figures
Similar articles
-
Induction of the Unfolded Protein Response during Bovine Alphaherpesvirus 1 Infection.Viruses. 2020 Sep 2;12(9):974. doi: 10.3390/v12090974. Viruses. 2020. PMID: 32887282 Free PMC article.
-
KSHV activates unfolded protein response sensors but suppresses downstream transcriptional responses to support lytic replication.PLoS Pathog. 2019 Dec 2;15(12):e1008185. doi: 10.1371/journal.ppat.1008185. eCollection 2019 Dec. PLoS Pathog. 2019. PMID: 31790507 Free PMC article.
-
The Human Cytomegalovirus Endoplasmic Reticulum-Resident Glycoprotein UL148 Activates the Unfolded Protein Response.J Virol. 2018 Sep 26;92(20):e00896-18. doi: 10.1128/JVI.00896-18. Print 2018 Oct 15. J Virol. 2018. PMID: 30045994 Free PMC article.
-
Shutoff of Host Gene Expression in Influenza A Virus and Herpesviruses: Similar Mechanisms and Common Themes.Viruses. 2016 Apr 16;8(4):102. doi: 10.3390/v8040102. Viruses. 2016. PMID: 27092522 Free PMC article. Review.
-
Coronavirus-induced ER stress response and its involvement in regulation of coronavirus-host interactions.Virus Res. 2014 Dec 19;194:110-23. doi: 10.1016/j.virusres.2014.09.016. Epub 2014 Oct 7. Virus Res. 2014. PMID: 25304691 Free PMC article. Review.
Cited by
-
Berberine in Human Oncogenic Herpesvirus Infections and Their Linked Cancers.Viruses. 2021 May 28;13(6):1014. doi: 10.3390/v13061014. Viruses. 2021. PMID: 34071559 Free PMC article. Review.
-
Induction of the Unfolded Protein Response during Bovine Alphaherpesvirus 1 Infection.Viruses. 2020 Sep 2;12(9):974. doi: 10.3390/v12090974. Viruses. 2020. PMID: 32887282 Free PMC article.
-
Multiple unfolded protein response pathways cooperate to link cytosolic dsDNA release to stimulator of interferon gene activation.Front Immunol. 2024 Jul 19;15:1358462. doi: 10.3389/fimmu.2024.1358462. eCollection 2024. Front Immunol. 2024. PMID: 39100663 Free PMC article.
-
Apoptosis during ZIKA Virus Infection: Too Soon or Too Late?Int J Mol Sci. 2022 Jan 24;23(3):1287. doi: 10.3390/ijms23031287. Int J Mol Sci. 2022. PMID: 35163212 Free PMC article. Review.
-
Viral mediated tethering to SEL1L facilitates ER-associated degradation of IRE1.J Virol. 2021 Mar 25;95(8):e01990-20. doi: 10.1128/JVI.01990-20. Epub 2021 Jan 20. J Virol. 2021. PMID: 33472927 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
