

Crossing fitness valleys via double substitutions within codons
- PMID: 31842858
- PMCID: PMC6916188
- DOI: 10.1186/s12915-019-0727-4
Crossing fitness valleys via double substitutions within codons
Abstract
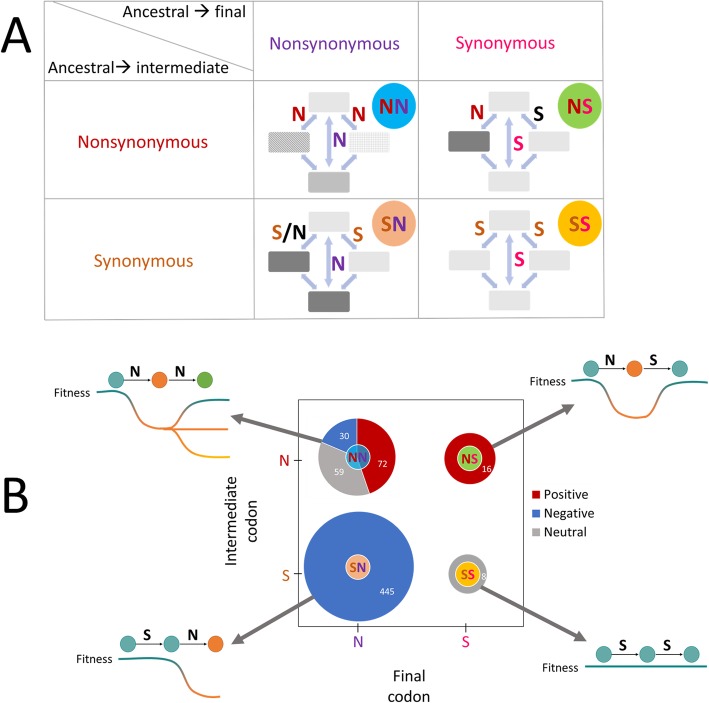
Background: Single nucleotide substitutions in protein-coding genes can be divided into synonymous (S), with little fitness effect, and non-synonymous (N) ones that alter amino acids and thus generally have a greater effect. Most of the N substitutions are affected by purifying selection that eliminates them from evolving populations. However, additional mutations of nearby bases potentially could alleviate the deleterious effect of single substitutions, making them subject to positive selection. To elucidate the effects of selection on double substitutions in all codons, it is critical to differentiate selection from mutational biases.
Results: We addressed the evolutionary regimes of within-codon double substitutions in 37 groups of closely related prokaryotic genomes from diverse phyla by comparing the fractions of double substitutions within codons to those of the equivalent double S substitutions in adjacent codons. Under the assumption that substitutions occur one at a time, all within-codon double substitutions can be represented as "ancestral-intermediate-final" sequences (where "intermediate" refers to the first single substitution and "final" refers to the second substitution) and can be partitioned into four classes: (1) SS, S intermediate-S final; (2) SN, S intermediate-N final; (3) NS, N intermediate-S final; and (4) NN, N intermediate-N final. We found that the selective pressure on the second substitution markedly differs among these classes of double substitutions. Analogous to single S (synonymous) substitutions, SS double substitutions evolve neutrally, whereas analogous to single N (non-synonymous) substitutions, SN double substitutions are subject to purifying selection. In contrast, NS show positive selection on the second step because the original amino acid is recovered. The NN double substitutions are heterogeneous and can be subject to either purifying or positive selection, or evolve neutrally, depending on the amino acid similarity between the final or intermediate and the ancestral states.
Conclusions: The results of the present, comprehensive analysis of the evolutionary landscape of within-codon double substitutions reaffirm the largely conservative regime of protein evolution. However, the second step of a double substitution can be subject to positive selection when the first step is deleterious. Such positive selection can result in frequent crossing of valleys on the fitness landscape.
Keywords: Archaea; Bacteria; DNA context; Double substitutions; Natural selection; Short-term evolution.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




Similar articles
-
Analysis of Stop Codons within Prokaryotic Protein-Coding Genes Suggests Frequent Readthrough Events.Int J Mol Sci. 2021 Feb 14;22(4):1876. doi: 10.3390/ijms22041876. Int J Mol Sci. 2021. PMID: 33672790 Free PMC article.
-
No evidence for widespread positive selection on double substitutions within codons in primates and yeasts.Front Genet. 2022 Sep 9;13:991249. doi: 10.3389/fgene.2022.991249. eCollection 2022. Front Genet. 2022. PMID: 36159983 Free PMC article.
-
Evolutionary switches between two serine codon sets are driven by selection.Proc Natl Acad Sci U S A. 2016 Nov 15;113(46):13109-13113. doi: 10.1073/pnas.1615832113. Epub 2016 Oct 31. Proc Natl Acad Sci U S A. 2016. PMID: 27799560 Free PMC article.
-
Extensive purifying selection acting on synonymous sites in HIV-1 Group M sequences.Virol J. 2008 Dec 23;5:160. doi: 10.1186/1743-422X-5-160. Virol J. 2008. PMID: 19105834 Free PMC article. Review.
-
DNA sequence evolution: the sounds of silence.Philos Trans R Soc Lond B Biol Sci. 1995 Sep 29;349(1329):241-7. doi: 10.1098/rstb.1995.0108. Philos Trans R Soc Lond B Biol Sci. 1995. PMID: 8577834 Review.
Cited by
-
Comparative transcriptomics of ice-crawlers demonstrates cold specialization constrains niche evolution in a relict lineage.Evol Appl. 2020 Sep 11;14(2):360-382. doi: 10.1111/eva.13120. eCollection 2021 Feb. Evol Appl. 2020. PMID: 33664782 Free PMC article.
-
Getting higher on rugged landscapes: Inversion mutations open access to fitter adaptive peaks in NK fitness landscapes.PLoS Comput Biol. 2022 Oct 31;18(10):e1010647. doi: 10.1371/journal.pcbi.1010647. eCollection 2022 Oct. PLoS Comput Biol. 2022. PMID: 36315581 Free PMC article.
-
Analysis of Stop Codons within Prokaryotic Protein-Coding Genes Suggests Frequent Readthrough Events.Int J Mol Sci. 2021 Feb 14;22(4):1876. doi: 10.3390/ijms22041876. Int J Mol Sci. 2021. PMID: 33672790 Free PMC article.
-
The 29-nucleotide deletion in SARS-CoV: truncated versions of ORF8 are under purifying selection.BMC Genomics. 2023 Jul 10;24(1):387. doi: 10.1186/s12864-023-09482-3. BMC Genomics. 2023. PMID: 37430204 Free PMC article.
-
No evidence for widespread positive selection on double substitutions within codons in primates and yeasts.Front Genet. 2022 Sep 9;13:991249. doi: 10.3389/fgene.2022.991249. eCollection 2022. Front Genet. 2022. PMID: 36159983 Free PMC article.
References
-
- Kimura M. The neutral theory of molecular evolution: Cambridge University Press. 1983.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
