

Impact of Suboptimal APOBEC3G Neutralization on the Emergence of HIV Drug Resistance in Humanized Mice
- PMID: 31801862
- PMCID: PMC7022346
- DOI: 10.1128/JVI.01543-19
Impact of Suboptimal APOBEC3G Neutralization on the Emergence of HIV Drug Resistance in Humanized Mice
Abstract
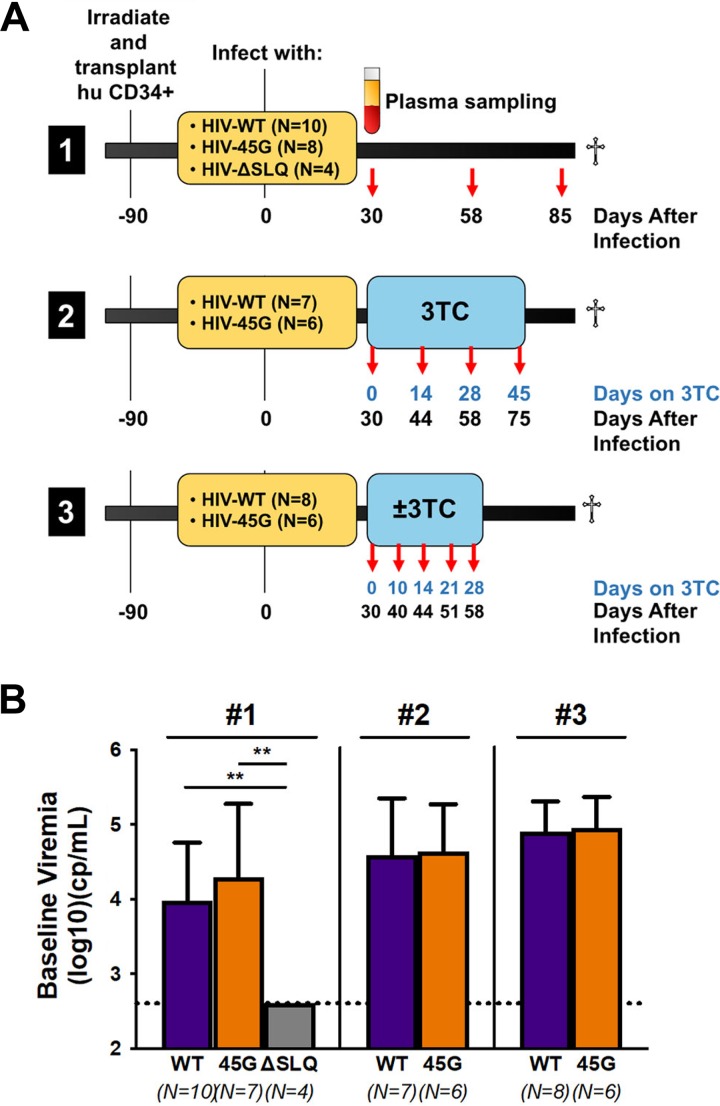
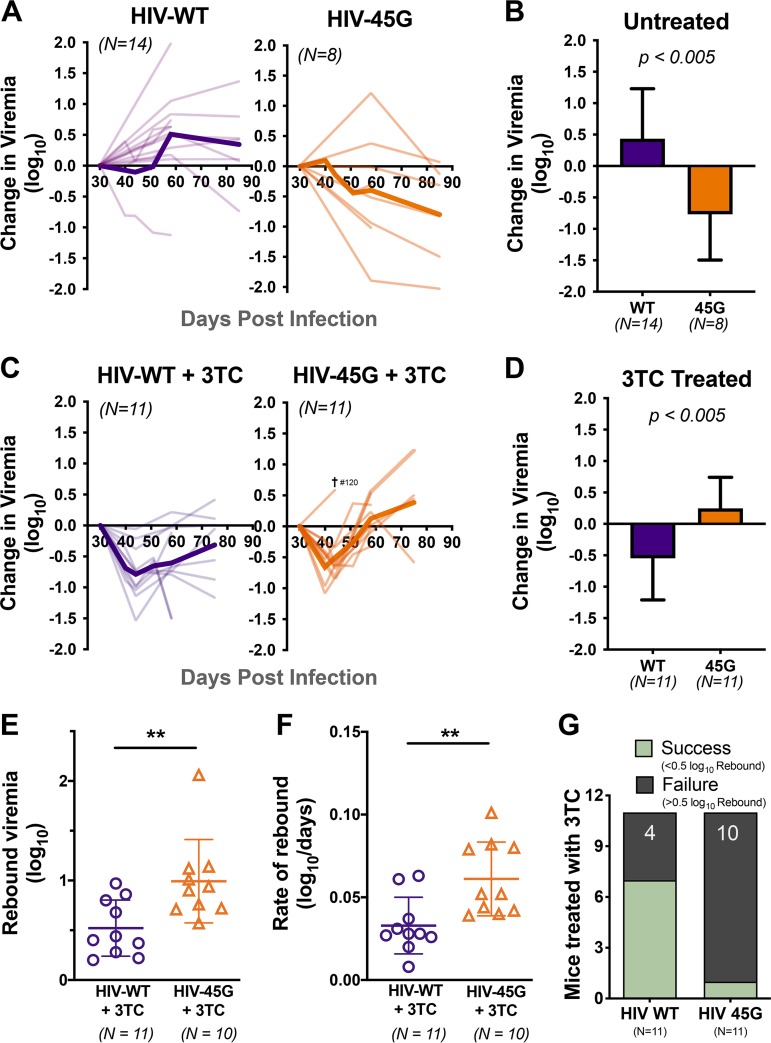
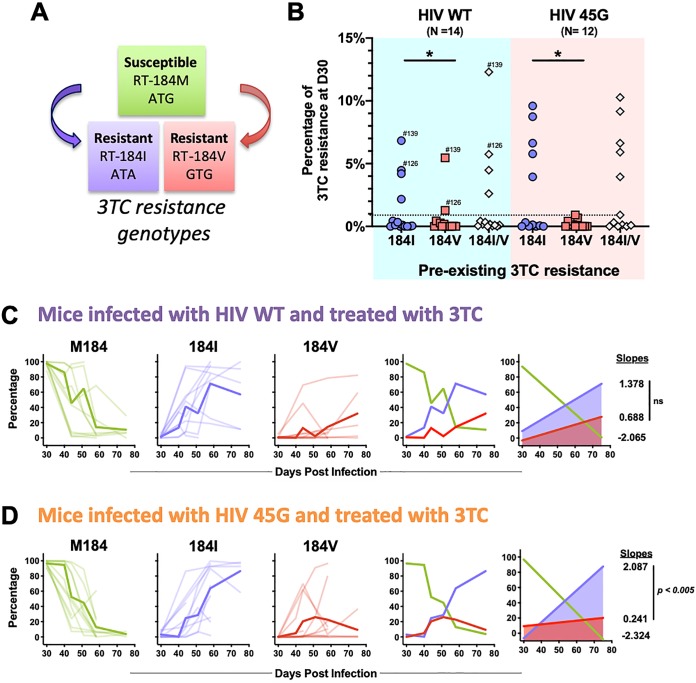
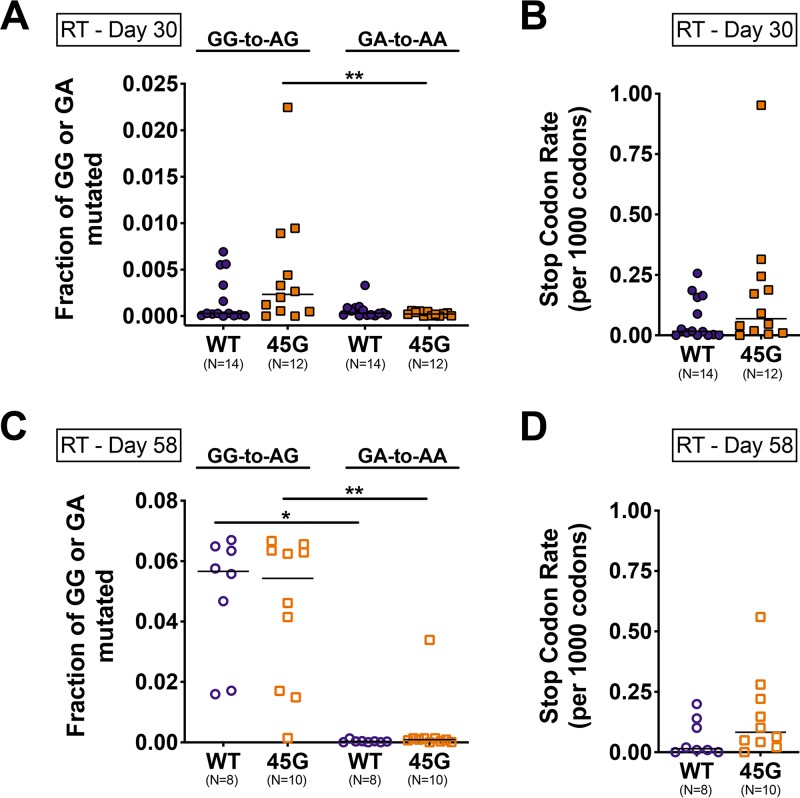
HIV diversification facilitates immune escape and complicates antiretroviral therapy. In this study, we take advantage of a humanized-mouse model to probe the contribution of APOBEC3 mutagenesis to viral evolution. Humanized mice were infected with isogenic HIV molecular clones (HIV-WT, HIV-45G, and HIV-ΔSLQ) that differ in their abilities to counteract APOBEC3G (A3G). Infected mice remained naive or were treated with the reverse transcriptase (RT) inhibitor lamivudine (3TC). Viremia, emergence of drug-resistant variants, and quasispecies diversification in the plasma compartment were determined throughout infection. While both HIV-WT and HIV-45G achieved robust infection, over time, HIV-45G replication was significantly reduced compared to that of HIV-WT in the absence of 3TC treatment. In contrast, treatment responses differed significantly between HIV-45G- and HIV-WT-infected mice. Antiretroviral treatment failed in 91% of HIV-45G-infected mice, while only 36% of HIV-WT-infected mice displayed a similar negative outcome. Emergence of 3TC-resistant variants and nucleotide diversity were determined by analyzing 155,462 single HIV reverse transcriptase gene (RT) and 6,985 vif sequences from 33 mice. Prior to treatment, variants with genotypic 3TC resistance (RT-M184I/V) were detected at low levels in over a third of all the animals. Upon treatment, the composition of the plasma quasispecies rapidly changed, leading to a majority of circulating viral variants encoding RT-184I. Interestingly, increased viral diversity prior to treatment initiation correlated with higher plasma viremia in HIV-45G-infected animals, but not in HIV-WT-infected animals. Taken together, HIV variants with suboptimal anti-A3G activity were attenuated in the absence of selection but displayed a fitness advantage in the presence of antiretroviral treatment.IMPORTANCE Both viral (e.g., RT) and host (e.g., A3G) factors can contribute to HIV sequence diversity. This study shows that suboptimal anti-A3G activity shapes viral fitness and drives viral evolution in the plasma compartment in humanized mice.
Keywords: APOBEC3; HIV Vif; HIV diversification; HIV drug resistance; human immunodeficiency virus; humanized mice; virus-host interactions.
Copyright © 2020 American Society for Microbiology.
Figures


Similar articles
-
Partially active HIV-1 Vif alleles facilitate viral escape from specific antiretrovirals.AIDS. 2010 Sep 24;24(15):2313-21. doi: 10.1097/QAD.0b013e32833e515a. AIDS. 2010. PMID: 20729708
-
APOBEC3G and APOBEC3F Act in Concert To Extinguish HIV-1 Replication.J Virol. 2016 Apr 14;90(9):4681-4695. doi: 10.1128/JVI.03275-15. Print 2016 May. J Virol. 2016. PMID: 26912618 Free PMC article.
-
The effect of HIV-1 Vif polymorphisms on A3G anti-viral activity in an in vivo mouse model.Retrovirology. 2016 Jun 30;13(1):45. doi: 10.1186/s12977-016-0280-y. Retrovirology. 2016. PMID: 27363431 Free PMC article.
-
Study of the impact of HIV genotypic drug resistance testing on therapy efficacy.Verh K Acad Geneeskd Belg. 2001;63(5):447-73. Verh K Acad Geneeskd Belg. 2001. PMID: 11813503 Review.
-
Apobec3G-Based Strategies to Defeat HIV Infection.Curr HIV Res. 2016;14(3):217-24. doi: 10.2174/1570162x14999160224100541. Curr HIV Res. 2016. PMID: 26957196 Review.
Cited by
-
Examination of the APOBEC3 Barrier to Cross Species Transmission of Primate Lentiviruses.Viruses. 2021 Jun 7;13(6):1084. doi: 10.3390/v13061084. Viruses. 2021. PMID: 34200141 Free PMC article. Review.
-
Small-Angle X-ray Scattering (SAXS) Measurements of APOBEC3G Provide Structural Basis for Binding of Single-Stranded DNA and Processivity.Viruses. 2022 Sep 6;14(9):1974. doi: 10.3390/v14091974. Viruses. 2022. PMID: 36146779 Free PMC article.
-
Next generation APOBEC3 inhibitors: Optimally designed for potency and nuclease stability.bioRxiv [Preprint]. 2024 Sep 6:2024.09.05.611238. doi: 10.1101/2024.09.05.611238. bioRxiv. 2024. PMID: 39282427 Free PMC article. Preprint.
-
APOBECs and Herpesviruses.Viruses. 2021 Feb 28;13(3):390. doi: 10.3390/v13030390. Viruses. 2021. PMID: 33671095 Free PMC article. Review.
-
Structural Insights into APOBEC3-Mediated Lentiviral Restriction.Viruses. 2020 May 27;12(6):587. doi: 10.3390/v12060587. Viruses. 2020. PMID: 32471198 Free PMC article. Review.
References
-
- Wood N, Bhattacharya T, Keele BF, Giorgi E, Liu M, Gaschen B, Daniels M, Ferrari G, Haynes BF, McMichael A, Shaw GM, Hahn BH, Korber B, Seoighe C. 2009. HIV evolution in early infection: selection pressures, patterns of insertion and deletion, and the impact of APOBEC. PLoS Pathog 5:e1000414. doi:10.1371/journal.ppat.1000414. - DOI - PMC - PubMed
-
- Rhodes TD, Nikolaitchik O, Chen J, Powell D, Hu WS. 2005. Genetic recombination of human immunodeficiency virus type 1 in one round of viral replication: effects of genetic distance, target cells, accessory genes, and lack of high negative interference in crossover events. J Virol 79:1666–1677. doi:10.1128/JVI.79.3.1666-1677.2005. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
