

A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C
- PMID: 31633398
- PMCID: PMC6803818
- DOI: 10.4155/fmc-2019-0181
A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C
Erratum in
-
Erratum.Future Med Chem. 2021 Mar;13(6):593-594. doi: 10.4155/fmc-2019-0181e1. Epub 2021 Feb 12. Future Med Chem. 2021. PMID: 33573418 Free PMC article. No abstract available.
Abstract
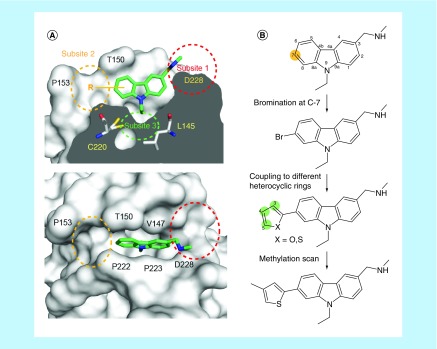
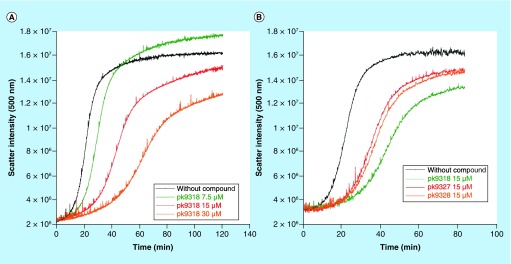
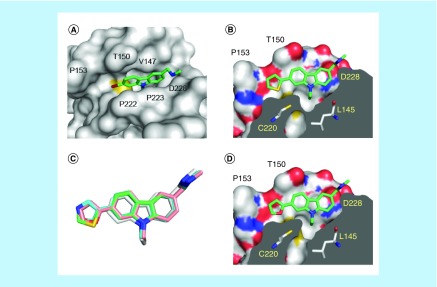
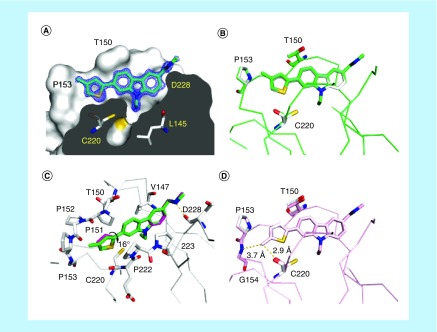
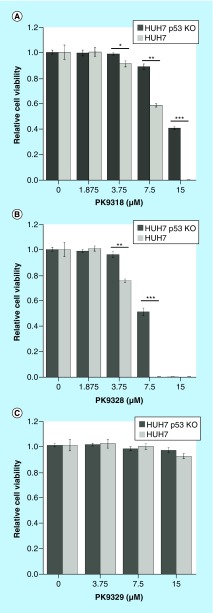
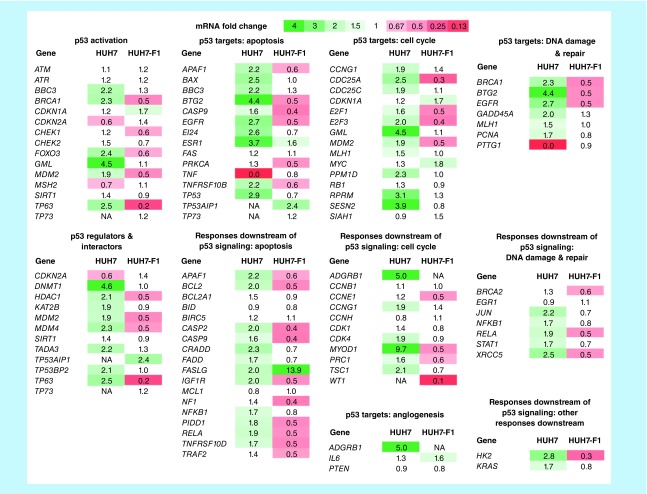
Aim: The p53 cancer mutation Y220C creates a conformationally unstable protein with a unique elongated surface crevice that can be targeted by molecular chaperones. We report the structure-guided optimization of the carbazole-based stabilizer PK083. Materials & methods: Biophysical, cellular and x-ray crystallographic techniques have been employed to elucidate the mode of action of the carbazole scaffolds. Results: Targeting an unoccupied subsite of the surface crevice with heterocycle-substituted PK083 analogs resulted in a 70-fold affinity increase to single-digit micromolar levels, increased thermal stability and decreased rate of aggregation of the mutant protein. PK9318, one of the most potent binders, restored p53 signaling in the liver cancer cell line HUH-7 with homozygous Y220C mutation. Conclusion: The p53-Y220C mutant is an excellent paradigm for the development of mutant p53 rescue drugs via protein stabilization. Similar rescue strategies may be applicable to other cavity-creating p53 cancer mutations.
Keywords: CRISPR/Cas9 p53 knockout; cancer mutations; cancer therapy; molecular chaperones; p53; protein stabilization; structure-based drug design; tumor suppression.
Conflict of interest statement
This work was funded by Worldwide Cancer Research (grants 14-1002, 18-0043), Sussex University (RN Jones), German Research Foundation (DFG) grant JO 1473/1-1 (AC Joerger) and ERC advanced grant 268506 (AR Fersht). The authors are grateful for support by the SGC, a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada through Ontario Genomics Institute, Innovative Medicines Initiative (EU/EFPIA) (ULTRA-DD grant no. 115766), Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and the Wellcome Trust.
No writing assistance was utilized in the production of this manuscript.
Figures
Similar articles
-
Targeting Cavity-Creating p53 Cancer Mutations with Small-Molecule Stabilizers: the Y220X Paradigm.ACS Chem Biol. 2020 Mar 20;15(3):657-668. doi: 10.1021/acschembio.9b00748. Epub 2020 Feb 21. ACS Chem Biol. 2020. PMID: 31990523 Free PMC article.
-
Virtual screening of p53 mutants reveals Y220S as an additional rescue drug target for PhiKan083 with higher binding characteristics.Comput Biol Chem. 2019 Jun;80:398-408. doi: 10.1016/j.compbiolchem.2019.05.005. Epub 2019 May 16. Comput Biol Chem. 2019. PMID: 31128451
-
Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines.Eur J Med Chem. 2018 May 25;152:101-114. doi: 10.1016/j.ejmech.2018.04.035. Epub 2018 Apr 21. Eur J Med Chem. 2018. PMID: 29702446 Free PMC article.
-
Mutant p53 reactivation by small molecules makes its way to the clinic.FEBS Lett. 2014 Aug 19;588(16):2622-7. doi: 10.1016/j.febslet.2014.04.017. Epub 2014 Apr 24. FEBS Lett. 2014. PMID: 24768524 Review.
-
Targeting the Prion-like Aggregation of Mutant p53 to Combat Cancer.Acc Chem Res. 2018 Jan 16;51(1):181-190. doi: 10.1021/acs.accounts.7b00473. Epub 2017 Dec 20. Acc Chem Res. 2018. PMID: 29260852 Review.
Cited by
-
Genotoxicity and Epigenotoxicity of Carbazole-Derived Molecules on MCF-7 Breast Cancer Cells.Int J Mol Sci. 2021 Mar 26;22(7):3410. doi: 10.3390/ijms22073410. Int J Mol Sci. 2021. PMID: 33810274 Free PMC article.
-
Therapeutic Strategies to Activate p53.Pharmaceuticals (Basel). 2022 Dec 24;16(1):24. doi: 10.3390/ph16010024. Pharmaceuticals (Basel). 2022. PMID: 36678521 Free PMC article. Review.
-
Advanced Strategies for Therapeutic Targeting of Wild-Type and Mutant p53 in Cancer.Biomolecules. 2022 Apr 6;12(4):548. doi: 10.3390/biom12040548. Biomolecules. 2022. PMID: 35454137 Free PMC article. Review.
-
Potential of rescue and reactivation of tumor suppressor p53 for cancer therapy.Biophys Rev. 2022 Jan 11;14(1):267-275. doi: 10.1007/s12551-021-00915-5. eCollection 2022 Feb. Biophys Rev. 2022. PMID: 35340607 Free PMC article. Review.
-
Targeting Mutant p53 for Cancer Treatment: Moving Closer to Clinical Use?Cancers (Basel). 2022 Sep 16;14(18):4499. doi: 10.3390/cancers14184499. Cancers (Basel). 2022. PMID: 36139658 Free PMC article. Review.
References
-
- Joerger AC, Fersht AR. The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu. Rev. Biochem. 85, 375–404 (2016). - PubMed
-
•• Comprehensive review of p53 drug discovery and structural biology as well as of the evolutionary history of p53 family proteins.
-
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell 137, 413–431 (2009). - PubMed
-
- Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 13, 217–236 (2014). - PubMed
-
•• Excellent review of different p53 drug discovery strategies.
-
- Bouaoun L, Sonkin D, Ardin M. et al. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum. Mutat. 37, 865–876 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous