

Proteome-level assessment of origin, prevalence and function of leucine-aspartic acid (LD) motifs
- PMID: 31584626
- PMCID: PMC7703752
- DOI: 10.1093/bioinformatics/btz703
Proteome-level assessment of origin, prevalence and function of leucine-aspartic acid (LD) motifs
Abstract
Motivation: Leucine-aspartic acid (LD) motifs are short linear interaction motifs (SLiMs) that link paxillin family proteins to factors controlling cell adhesion, motility and survival. The existence and importance of LD motifs beyond the paxillin family is poorly understood.
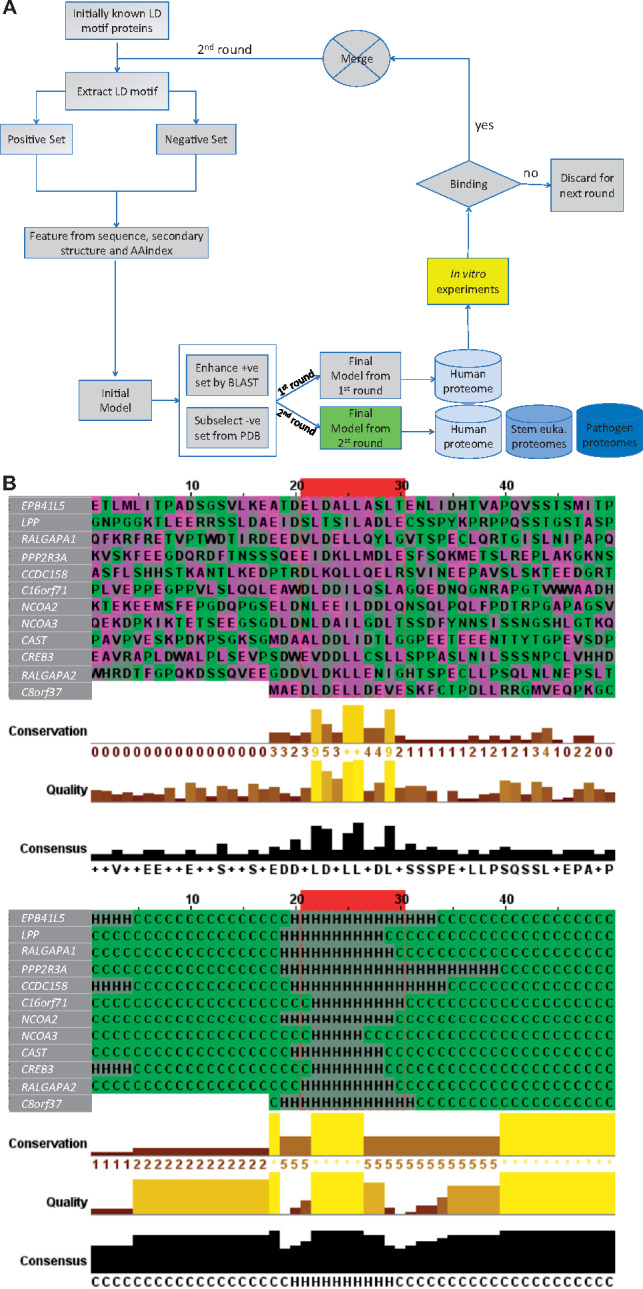
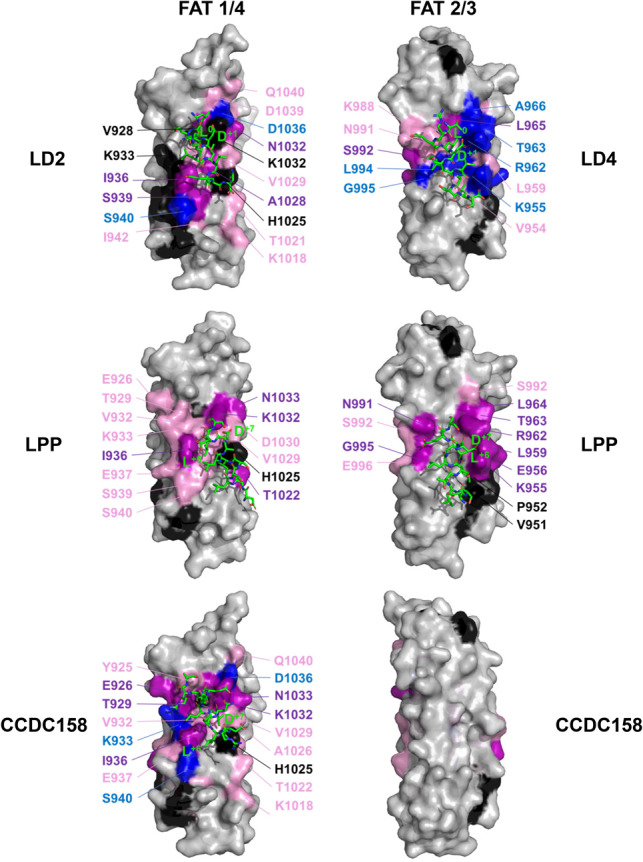
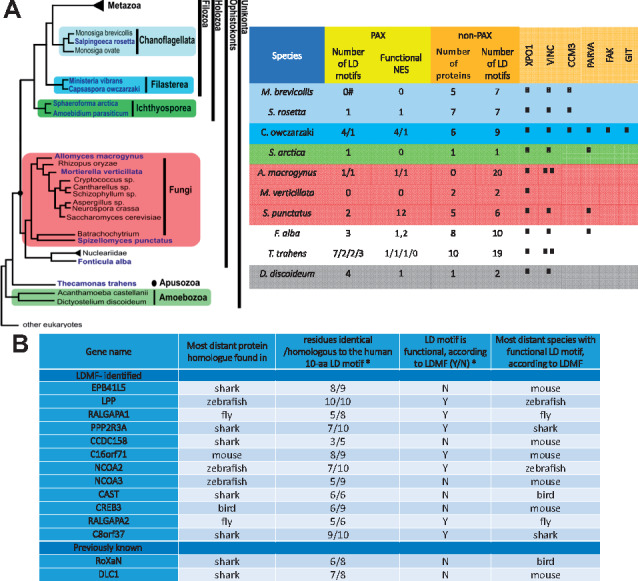
Results: To enable a proteome-wide assessment of LD motifs, we developed an active learning based framework (LD motif finder; LDMF) that iteratively integrates computational predictions with experimental validation. Our analysis of the human proteome revealed a dozen new proteins containing LD motifs. We found that LD motif signalling evolved in unicellular eukaryotes more than 800 Myr ago, with paxillin and vinculin as core constituents, and nuclear export signal as a likely source of de novo LD motifs. We show that LD motif proteins form a functionally homogenous group, all being involved in cell morphogenesis and adhesion. This functional focus is recapitulated in cells by GFP-fused LD motifs, suggesting that it is intrinsic to the LD motif sequence, possibly through their effect on binding partners. Our approach elucidated the origin and dynamic adaptations of an ancestral SLiM, and can serve as a guide for the identification of other SLiMs for which only few representatives are known.
Availability and implementation: LDMF is freely available online at www.cbrc.kaust.edu.sa/ldmf; Source code is available at https://github.com/tanviralambd/LD/.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2019. Published by Oxford University Press.
Figures



Similar articles
-
How to find a leucine in a haystack? Structure, ligand recognition and regulation of leucine-aspartic acid (LD) motifs.Biochem J. 2014 Jun 15;460(3):317-29. doi: 10.1042/BJ20140298. Biochem J. 2014. PMID: 24870021 Review.
-
Spatial arrangement of LD motif-interacting residues on focal adhesion targeting domain of Focal Adhesion Kinase determine domain-motif interaction affinity and specificity.Biochim Biophys Acta Gen Subj. 2020 Jan;1864(1):129450. doi: 10.1016/j.bbagen.2019.129450. Epub 2019 Oct 30. Biochim Biophys Acta Gen Subj. 2020. PMID: 31676296
-
Bioinformatics Approaches for Predicting Disordered Protein Motifs.Adv Exp Med Biol. 2015;870:291-318. doi: 10.1007/978-3-319-20164-1_9. Adv Exp Med Biol. 2015. PMID: 26387106 Review.
-
Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: A role in cytoskeletal remodeling.J Cell Biol. 1999 May 17;145(4):851-63. doi: 10.1083/jcb.145.4.851. J Cell Biol. 1999. PMID: 10330411 Free PMC article.
-
SLiMSearch: a framework for proteome-wide discovery and annotation of functional modules in intrinsically disordered regions.Nucleic Acids Res. 2017 Jul 3;45(W1):W464-W469. doi: 10.1093/nar/gkx238. Nucleic Acids Res. 2017. PMID: 28387819 Free PMC article.
Cited by
-
Endogenous Control Mechanisms of FAK and PYK2 and Their Relevance to Cancer Development.Cancers (Basel). 2018 Jun 11;10(6):196. doi: 10.3390/cancers10060196. Cancers (Basel). 2018. PMID: 29891810 Free PMC article. Review.
-
The structural basis of the talin-KANK1 interaction that coordinates the actin and microtubule cytoskeletons at focal adhesions.Open Biol. 2023 Jun;13(6):230058. doi: 10.1098/rsob.230058. Epub 2023 Jun 21. Open Biol. 2023. PMID: 37339751 Free PMC article.
-
Paxillin: A Hub for Mechano-Transduction from the β3 Integrin-Talin-Kindlin Axis.Front Cell Dev Biol. 2022 Apr 5;10:852016. doi: 10.3389/fcell.2022.852016. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 35450290 Free PMC article. Review.
-
Deep Learning in LncRNAome: Contribution, Challenges, and Perspectives.Noncoding RNA. 2020 Nov 30;6(4):47. doi: 10.3390/ncrna6040047. Noncoding RNA. 2020. PMID: 33266128 Free PMC article. Review.
-
miRCOVID-19: Potential Targets of Human miRNAs in SARS-CoV-2 for RNA-Based Drug Discovery.Noncoding RNA. 2021 Mar 2;7(1):18. doi: 10.3390/ncrna7010018. Noncoding RNA. 2021. PMID: 33801496 Free PMC article.
References
-
- Alam T. et al. (2014) How to find a leucine in a haystack? Structure, ligand recognition and regulation of leucine-aspartic acid (LD) motifs. Biochem. J., 460, 317–329. - PubMed
-
- Arold S.T. et al. (2002) The structural basis of localization and signaling by the focal adhesion targeting domain. Structure, 10, 319–327. - PubMed
-
- Astro V. et al. (2011) Liprin-alpha1 regulates breast cancer cell invasion by affecting cell motility, invadopodia and extracellular matrix degradation. Oncogene, 30, 1841–1849. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
