

Apolipoprotein AI) Promotes Atherosclerosis Regression in Diabetic Mice by Suppressing Myelopoiesis and Plaque Inflammation
- PMID: 31567014
- PMCID: PMC6777860
- DOI: 10.1161/CIRCULATIONAHA.119.039476
Apolipoprotein AI) Promotes Atherosclerosis Regression in Diabetic Mice by Suppressing Myelopoiesis and Plaque Inflammation
Abstract
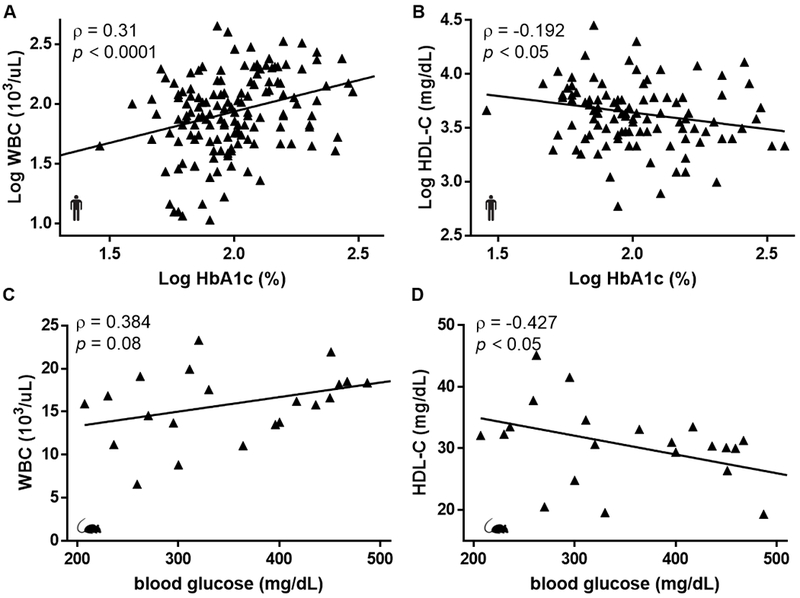
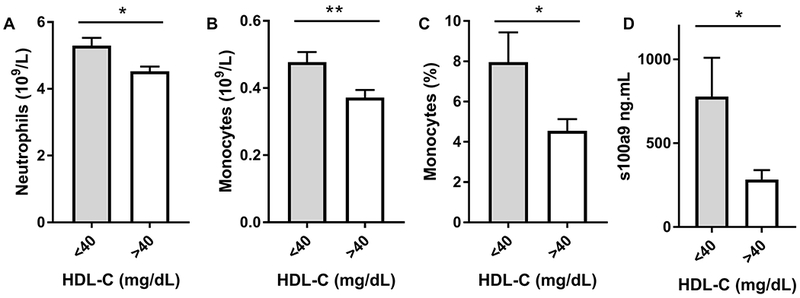
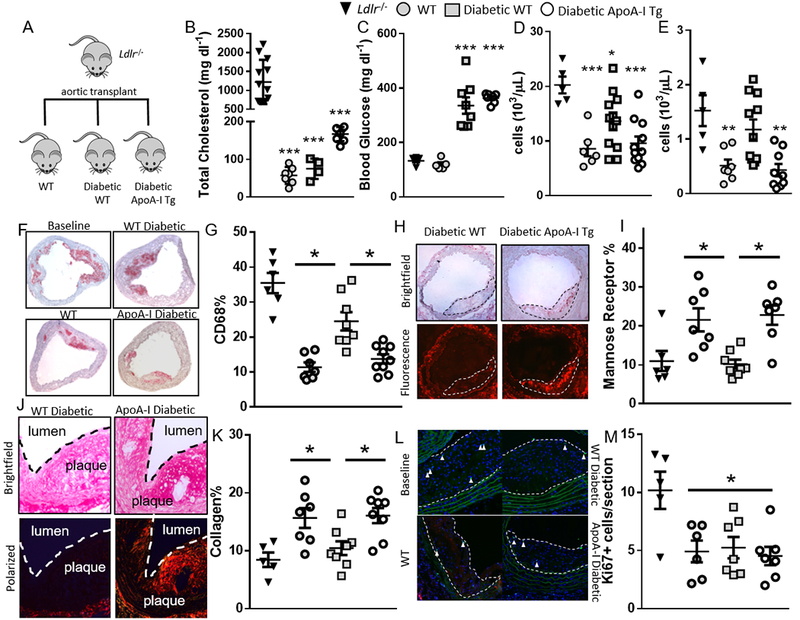
Background: Despite robust cholesterol lowering, cardiovascular disease risk remains increased in patients with diabetes mellitus. Consistent with this, diabetes mellitus impairs atherosclerosis regression after cholesterol lowering in humans and mice. In mice, this is attributed in part to hyperglycemia-induced monocytosis, which increases monocyte entry into plaques despite cholesterol lowering. In addition, diabetes mellitus skews plaque macrophages toward an atherogenic inflammatory M1 phenotype instead of toward the atherosclerosis-resolving M2 state typical with cholesterol lowering. Functional high-density lipoprotein (HDL), typically low in patients with diabetes mellitus, reduces monocyte precursor proliferation in murine bone marrow and has anti-inflammatory effects on human and murine macrophages. Our study aimed to test whether raising functional HDL levels in diabetic mice prevents monocytosis, reduces the quantity and inflammation of plaque macrophages, and enhances atherosclerosis regression after cholesterol lowering.
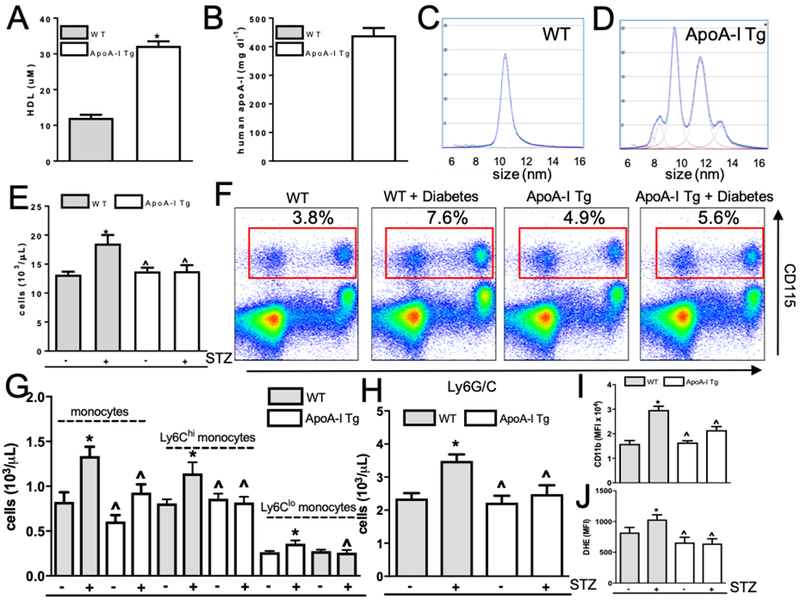
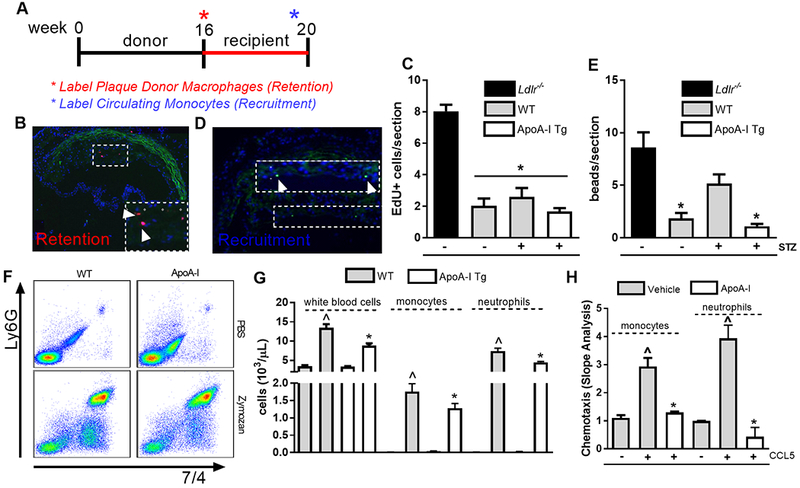
Methods: Aortic arches containing plaques developed in Ldlr-/- mice were transplanted into either wild-type, diabetic wild-type, or diabetic mice transgenic for human apolipoprotein AI, which have elevated functional HDL. Recipient mice all had low levels of low-density lipoprotein cholesterol to promote plaque regression. After 2 weeks, plaques in recipient mouse aortic grafts were examined.
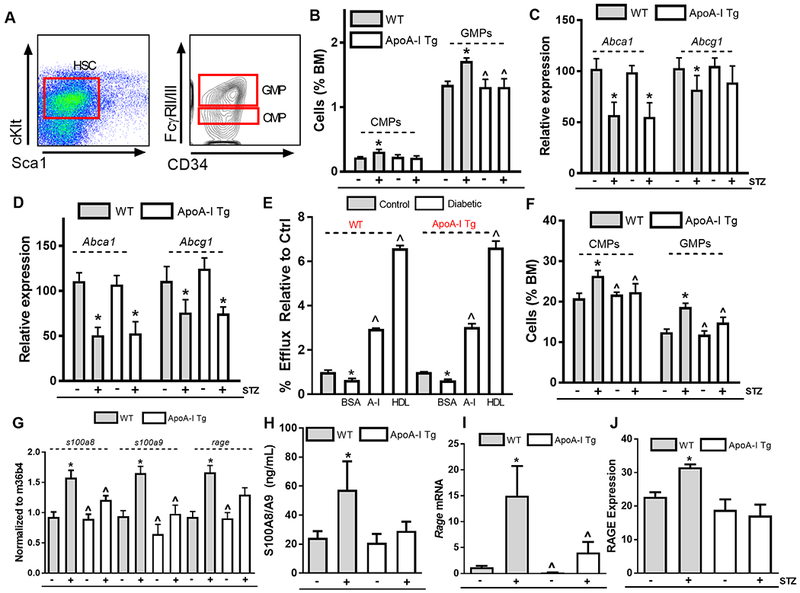
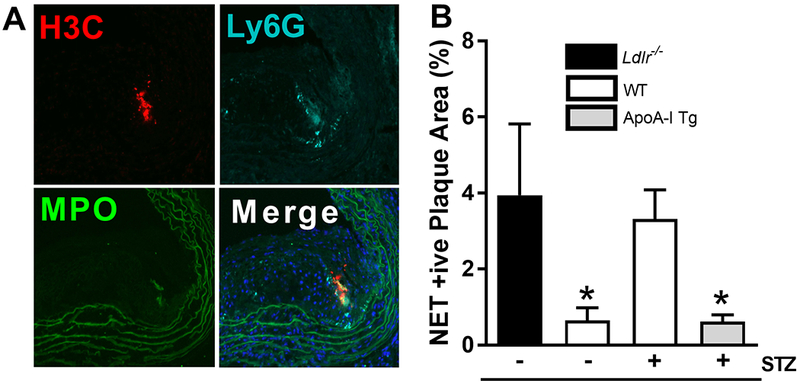
Results: Diabetic wild-type mice had impaired atherosclerosis regression, which was normalized by raising HDL levels. This benefit was linked to suppressed hyperglycemia-driven myelopoiesis, monocytosis, and neutrophilia. Increased HDL improved cholesterol efflux from bone marrow progenitors, suppressing their proliferation and monocyte and neutrophil production capacity. In addition to reducing circulating monocytes available for recruitment into plaques, in the diabetic milieu, HDL suppressed the general recruitability of monocytes to inflammatory sites and promoted plaque macrophage polarization to the M2, atherosclerosis-resolving state. There was also a decrease in plaque neutrophil extracellular traps, which are atherogenic and increased by diabetes mellitus.
Conclusions: Raising apolipoprotein AI and functional levels of HDL promotes multiple favorable changes in the production of monocytes and neutrophils and in the inflammatory environment of atherosclerotic plaques of diabetic mice after cholesterol lowering and may represent a novel approach to reduce cardiovascular disease risk in people with diabetes mellitus.
Keywords: apolipoproteins; arteriosclerosis; diabetes mellitus; leukocytosis; lipoproteins, HDL; macrophages; myelopoiesis.
Figures
Comment in
-
Enhancing Atherosclerosis Regression in Diabetic Mice Through Apo AI (Apolipoprotein AI).Circulation. 2019 Oct;140(14):1185-1187. doi: 10.1161/CIRCULATIONAHA.119.040369. Epub 2019 Sep 30. Circulation. 2019. PMID: 31567013 No abstract available.
Similar articles
-
miR33 inhibition overcomes deleterious effects of diabetes mellitus on atherosclerosis plaque regression in mice.Circ Res. 2014 Oct 10;115(9):759-69. doi: 10.1161/CIRCRESAHA.115.304164. Epub 2014 Sep 8. Circ Res. 2014. PMID: 25201910 Free PMC article.
-
SGLT2 inhibition reduces atherosclerosis by enhancing lipoprotein clearance in Ldlr-/- type 1 diabetic mice.Atherosclerosis. 2018 Apr;271:166-176. doi: 10.1016/j.atherosclerosis.2018.02.028. Epub 2018 Mar 2. Atherosclerosis. 2018. PMID: 29518749 Free PMC article.
-
Imbalance of APOB Lipoproteins and Large HDL in Type 1 Diabetes Drives Atherosclerosis.Circ Res. 2024 Jul 5;135(2):335-349. doi: 10.1161/CIRCRESAHA.123.323100. Epub 2024 Jun 3. Circ Res. 2024. PMID: 38828596 Free PMC article.
-
Cholesterol efflux: a novel regulator of myelopoiesis and atherogenesis.Arterioscler Thromb Vasc Biol. 2012 Nov;32(11):2547-52. doi: 10.1161/ATVBAHA.112.300134. Arterioscler Thromb Vasc Biol. 2012. PMID: 23077140 Review.
-
Anti-atherogenic mechanisms of high density lipoprotein: effects on myeloid cells.Biochim Biophys Acta. 2012 Mar;1821(3):513-21. doi: 10.1016/j.bbalip.2011.08.003. Epub 2011 Aug 16. Biochim Biophys Acta. 2012. PMID: 21864714 Free PMC article. Review.
Cited by
-
CRISPR/dCas9 Transcriptional Activation of Endogenous Apolipoprotein AI and Paraoxonase 1 in Enterocytes Alleviates Endothelial Cell Dysfunction.Biomolecules. 2021 Nov 25;11(12):1769. doi: 10.3390/biom11121769. Biomolecules. 2021. PMID: 34944413 Free PMC article.
-
Expression of recombinant human Apolipoprotein A-IMilano in Nicotiana tabacum.Bioresour Bioprocess. 2023 Jan 21;10(1):4. doi: 10.1186/s40643-023-00623-w. Bioresour Bioprocess. 2023. PMID: 38647895 Free PMC article.
-
β-Carotene accelerates the resolution of atherosclerosis in mice.Elife. 2024 Feb 6;12:RP87430. doi: 10.7554/eLife.87430. Elife. 2024. PMID: 38319073 Free PMC article.
-
The purine metabolite inosine monophosphate accelerates myelopoiesis and acute pancreatitis progression.Commun Biol. 2022 Oct 12;5(1):1088. doi: 10.1038/s42003-022-04041-0. Commun Biol. 2022. PMID: 36224248 Free PMC article.
-
Curcuminoid supplementation in canine diabetic mellitus and its complications using proteomic analysis.Front Vet Sci. 2022 Dec 23;9:1057972. doi: 10.3389/fvets.2022.1057972. eCollection 2022. Front Vet Sci. 2022. PMID: 36619946 Free PMC article.
References
-
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K and Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. New Eng J Med. 1998;339:229–234. - PubMed
-
- Cholesterol Treatment Trialists C, Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. - PMC - PubMed
-
- Hiro T, Kimura T, Morimoto T, Miyauchi K, Nakagawa Y, Yamagishi M, Ozaki Y, Kimura K, Saito S, Yamaguchi T, et al. Diabetes mellitus is a major negative determinant of coronary plaque regression during statin therapy in patients with acute coronary syndrome--serial intravascular ultrasound observations from the Japan Assessment of Pitavastatin and Atorvastatin in Acute Coronary Syndrome Trial (the JAPAN-ACS Trial). Circulation journal : official journal of the Japanese Circulation Society. 2010;74:1165–1174. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL117226/HL/NHLBI NIH HHS/United States
- R01 HL114978/HL/NHLBI NIH HHS/United States
- P01 HL131481/HL/NHLBI NIH HHS/United States
- R01 HL045095/HL/NHLBI NIH HHS/United States
- R01 HL129433/HL/NHLBI NIH HHS/United States
- DP3 DK108209/DK/NIDDK NIH HHS/United States
- R01 HL139909/HL/NHLBI NIH HHS/United States
- P01 HL092969/HL/NHLBI NIH HHS/United States
- R01 DK095684/DK/NIDDK NIH HHS/United States
- P30 DK017047/DK/NIDDK NIH HHS/United States
- R01 HL084312/HL/NHLBI NIH HHS/United States
- R35 HL144993/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
