

Precocious chondrocyte differentiation disrupts skeletal growth in Kabuki syndrome mice
- PMID: 31557133
- PMCID: PMC6824315
- DOI: 10.1172/jci.insight.129380
Precocious chondrocyte differentiation disrupts skeletal growth in Kabuki syndrome mice
Abstract
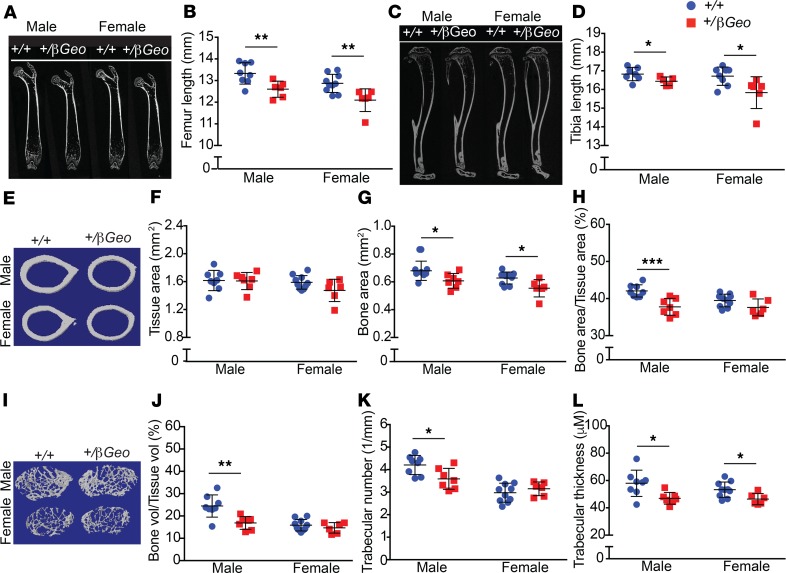
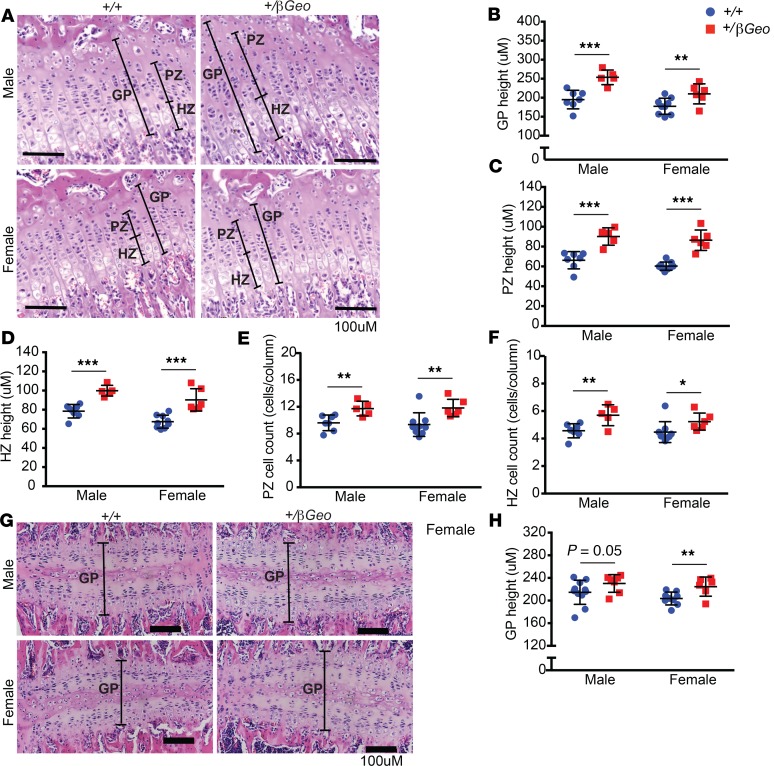
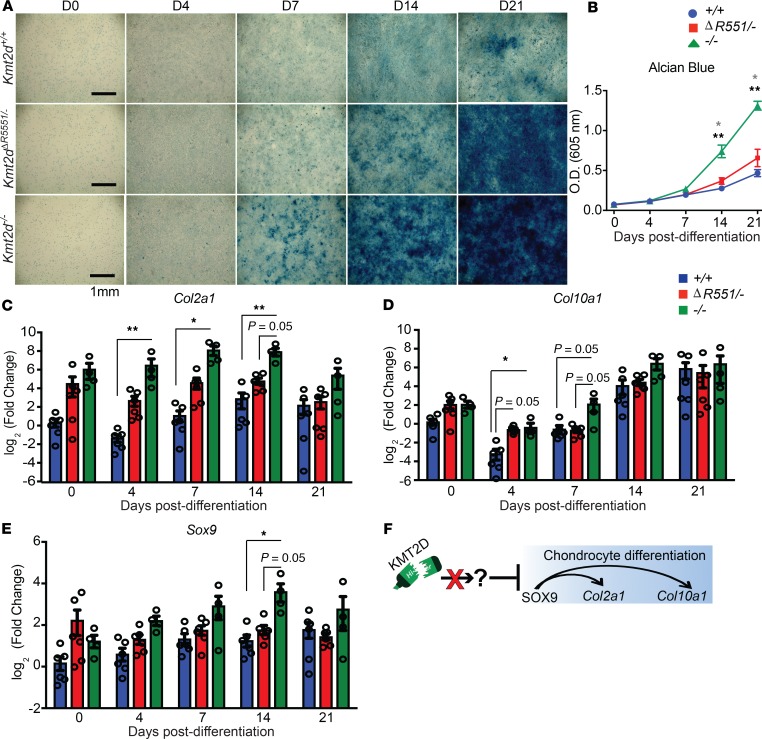
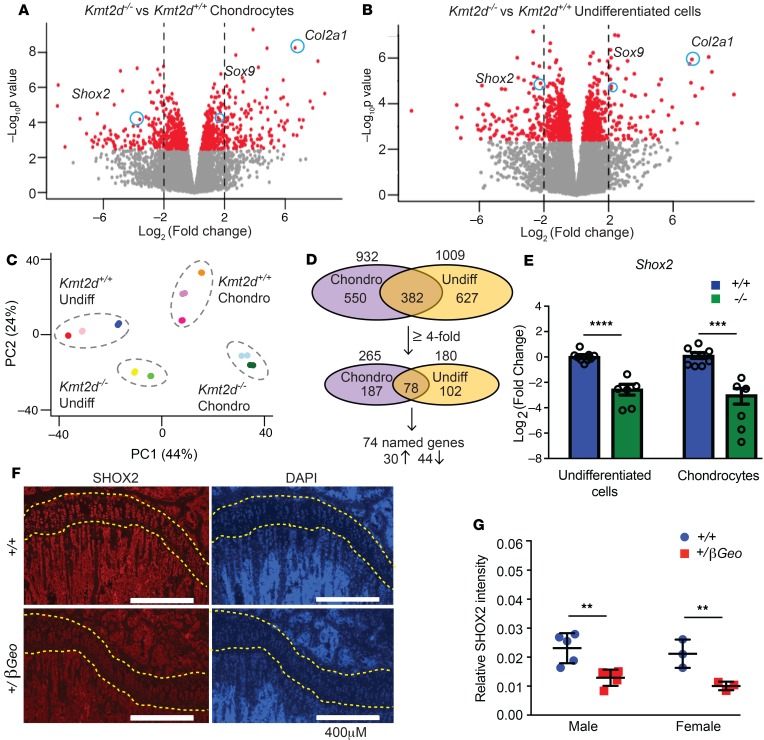
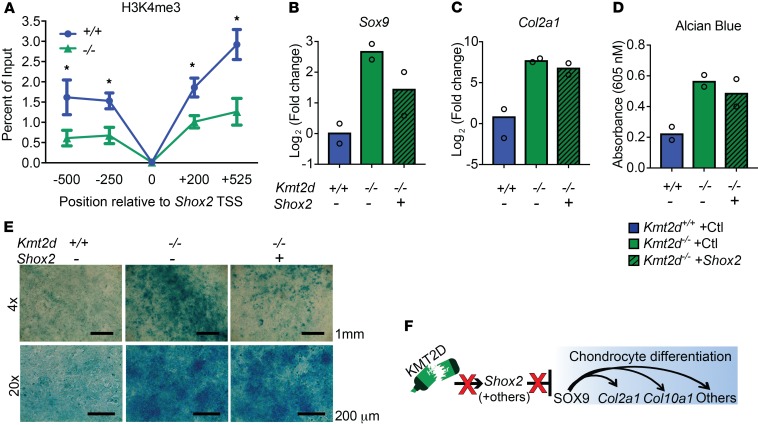
Kabuki syndrome 1 (KS1) is a Mendelian disorder of the epigenetic machinery caused by mutations in the gene encoding KMT2D, which methylates lysine 4 on histone H3 (H3K4). KS1 is characterized by intellectual disability, postnatal growth retardation, and distinct craniofacial dysmorphisms. A mouse model (Kmt2d+/βGeo) exhibits features of the human disorder and has provided insight into other phenotypes; however, the mechanistic basis of skeletal abnormalities and growth retardation remains elusive. Using high-resolution micro-CT, we show that Kmt2d+/βGeo mice have shortened long bones and ventral bowing of skulls. In vivo expansion of growth plates within skulls and long bones suggests disrupted endochondral ossification as a common disease mechanism. Stable chondrocyte cell lines harboring inactivating mutations in Kmt2d exhibit precocious differentiation, further supporting this mechanism. A known inducer of chondrogenesis, SOX9, and its targets show markedly increased expression in Kmt2d-/- chondrocytes. By transcriptome profiling, we identify Shox2 as a putative KMT2D target. We propose that decreased KMT2D-mediated H3K4me3 at Shox2 releases Sox9 inhibition and thereby leads to enhanced chondrogenesis, providing a potentially novel and plausible explanation for precocious chondrocyte differentiation. Our findings provide insight into the pathogenesis of growth retardation in KS1 and suggest therapeutic approaches for this and related disorders.
Keywords: Epigenetics; Genetic diseases; Genetics.
Conflict of interest statement
Figures

Similar articles
-
The KMT2D Kabuki syndrome histone methylase controls neural crest cell differentiation and facial morphology.Development. 2020 Jul 17;147(21):dev187997. doi: 10.1242/dev.187997. Development. 2020. PMID: 32541010 Free PMC article.
-
Precocious neuronal differentiation and disrupted oxygen responses in Kabuki syndrome.JCI Insight. 2019 Oct 17;4(20):e129375. doi: 10.1172/jci.insight.129375. JCI Insight. 2019. PMID: 31465303 Free PMC article.
-
Growth deficiency in a mouse model of Kabuki syndrome 2 bears mechanistic similarities to Kabuki syndrome 1.PLoS Genet. 2024 Jun 10;20(6):e1011310. doi: 10.1371/journal.pgen.1011310. eCollection 2024 Jun. PLoS Genet. 2024. PMID: 38857303 Free PMC article.
-
Using Xenopus to analyze neurocristopathies like Kabuki syndrome.Genesis. 2021 Feb;59(1-2):e23404. doi: 10.1002/dvg.23404. Epub 2020 Dec 22. Genesis. 2021. PMID: 33351273 Review.
-
Kabuki syndrome: novel pathogenic variants, new phenotypes and review of literature.Orphanet J Rare Dis. 2019 Nov 14;14(1):255. doi: 10.1186/s13023-019-1219-x. Orphanet J Rare Dis. 2019. PMID: 31727177 Free PMC article. Review.
Cited by
-
Are Copy Number Variations within the FecB Gene Significantly Associated with Morphometric Traits in Goats?Animals (Basel). 2022 Jun 15;12(12):1547. doi: 10.3390/ani12121547. Animals (Basel). 2022. PMID: 35739883 Free PMC article.
-
Lysine methyltransferase 2D regulates muscle fiber size and muscle cell differentiation.FASEB J. 2021 Nov;35(11):e21955. doi: 10.1096/fj.202100823R. FASEB J. 2021. PMID: 34613626 Free PMC article.
-
Expanding the genotypic and phenotypic spectrum in a diverse cohort of 104 individuals with Wiedemann-Steiner syndrome.Am J Med Genet A. 2021 Jun;185(6):1649-1665. doi: 10.1002/ajmg.a.62124. Epub 2021 Mar 30. Am J Med Genet A. 2021. PMID: 33783954 Free PMC article.
-
The KMT2D Kabuki syndrome histone methylase controls neural crest cell differentiation and facial morphology.Development. 2020 Jul 17;147(21):dev187997. doi: 10.1242/dev.187997. Development. 2020. PMID: 32541010 Free PMC article.
-
Leveraging the Mendelian disorders of the epigenetic machinery to systematically map functional epigenetic variation.Elife. 2021 Aug 31;10:e65884. doi: 10.7554/eLife.65884. Elife. 2021. PMID: 34463256 Free PMC article.
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
