

Depalmitoylation by Palmitoyl-Protein Thioesterase 1 in Neuronal Health and Degeneration
- PMID: 31555119
- PMCID: PMC6727029
- DOI: 10.3389/fnsyn.2019.00025
Depalmitoylation by Palmitoyl-Protein Thioesterase 1 in Neuronal Health and Degeneration
Abstract
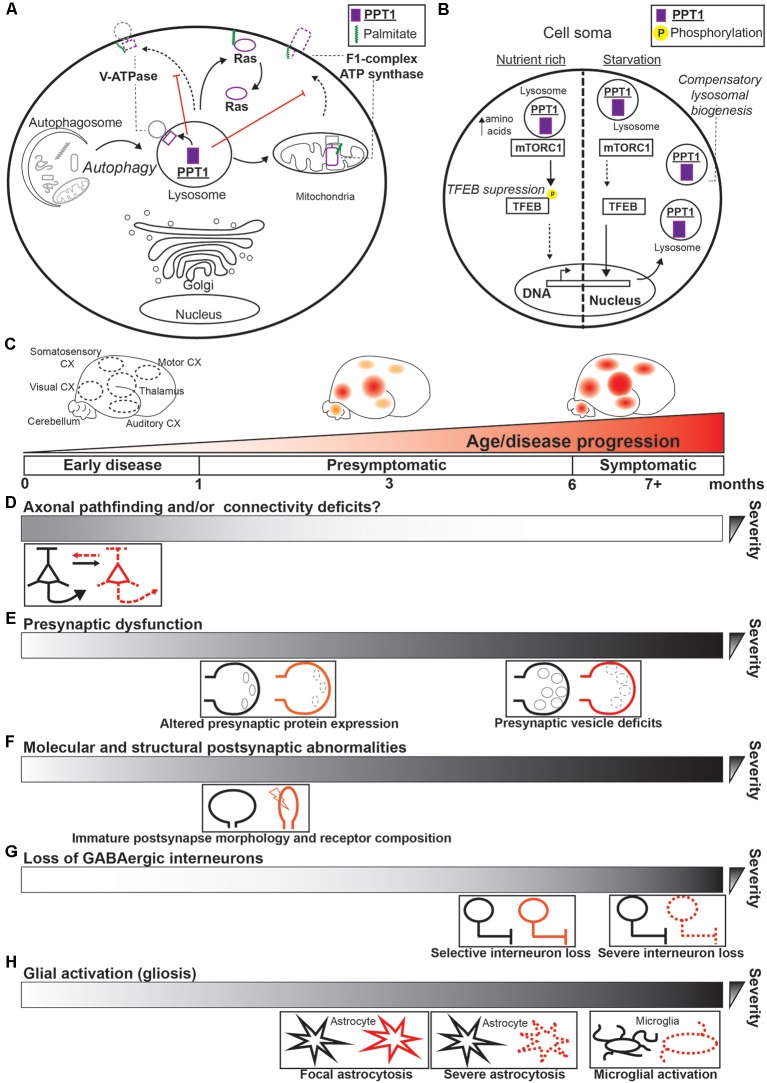
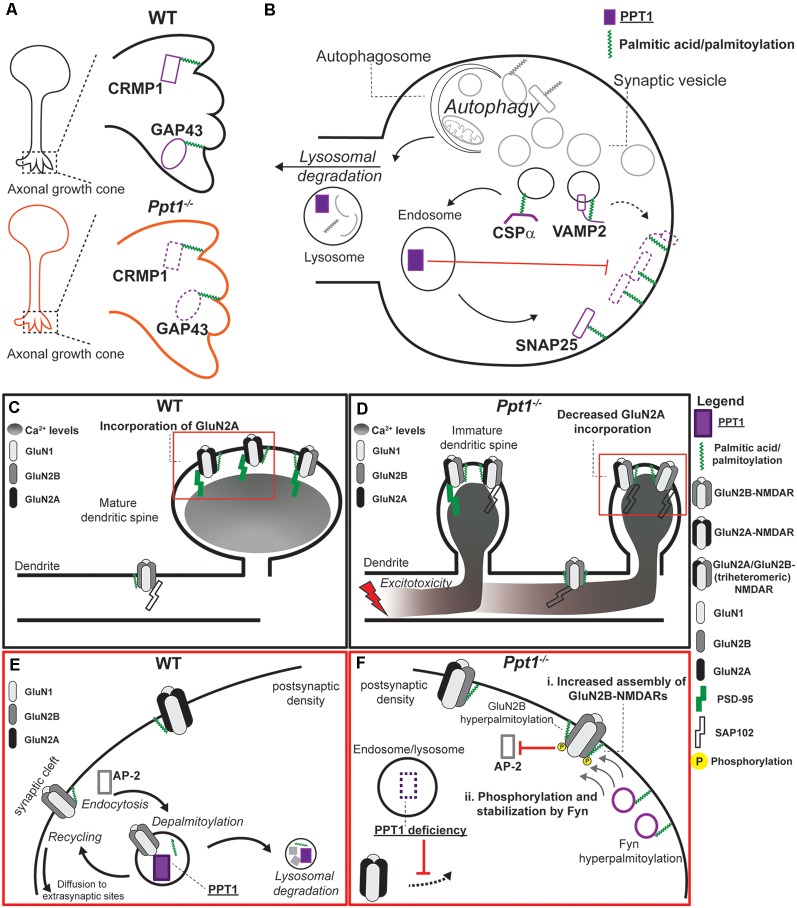
Protein palmitoylation is the post-translational, reversible addition of a 16-carbon fatty acid, palmitate, to proteins. Protein palmitoylation has recently garnered much attention, as it robustly modifies the localization and function of canonical signaling molecules and receptors. Protein depalmitoylation, on the other hand, is the process by which palmitic acid is removed from modified proteins and contributes, therefore, comparably to palmitoylated-protein dynamics. Palmitoylated proteins also require depalmitoylation prior to lysosomal degradation, demonstrating the significance of this process in protein sorting and turnover. Palmitoylation and depalmitoylation serve as particularly crucial regulators of protein function in neurons, where a specialized molecular architecture and cholesterol-rich membrane microdomains contribute to synaptic transmission. Three classes of depalmitoylating enzymes are currently recognized, the acyl protein thioesterases, α/β hydrolase domain-containing 17 proteins (ABHD17s), and the palmitoyl-protein thioesterases (PPTs). However, a clear picture of depalmitoylation has not yet emerged, in part because the enzyme-substrate relationships and specific functions of depalmitoylation are only beginning to be uncovered. Further, despite the finding that loss-of-function mutations affecting palmitoyl-protein thioesterase 1 (PPT1) function cause a severe pediatric neurodegenerative disease, the role of PPT1 as a depalmitoylase has attracted relatively little attention. Understanding the role of depalmitoylation by PPT1 in neuronal function is a fertile area for ongoing basic science and translational research that may have broader therapeutic implications for neurodegeneration. Here, we will briefly introduce the rapidly growing field surrounding protein palmitoylation and depalmitoylation, then will focus on the role of PPT1 in development, health, and neurological disease.
Keywords: NMDA; PPT1; depalmitoylation; lipofuscinosis; neurodegeneration; palmitoylation.
Figures
Similar articles
-
Loss of Depalmitoylation Disrupts Homeostatic Plasticity of AMPARs in a Mouse Model of Infantile Neuronal Ceroid Lipofuscinosis.J Neurosci. 2023 Dec 6;43(49):8317-8335. doi: 10.1523/JNEUROSCI.1113-23.2023. J Neurosci. 2023. PMID: 37884348 Free PMC article.
-
Identification of substrates of palmitoyl protein thioesterase 1 highlights roles of depalmitoylation in disulfide bond formation and synaptic function.PLoS Biol. 2022 Mar 31;20(3):e3001590. doi: 10.1371/journal.pbio.3001590. eCollection 2022 Mar. PLoS Biol. 2022. PMID: 35358180 Free PMC article.
-
Reversible Cysteine Acylation Regulates the Activity of Human Palmitoyl-Protein Thioesterase 1 (PPT1).PLoS One. 2016 Jan 5;11(1):e0146466. doi: 10.1371/journal.pone.0146466. eCollection 2016. PLoS One. 2016. PMID: 26731412 Free PMC article.
-
Protein palmitoylation in cancer: molecular functions and therapeutic potential.Mol Oncol. 2023 Jan;17(1):3-26. doi: 10.1002/1878-0261.13308. Epub 2022 Sep 10. Mol Oncol. 2023. PMID: 36018061 Free PMC article. Review.
-
Protein Palmitoylation by DHHC Protein Family.In: Kittler JT, Moss SJ, editors. The Dynamic Synapse: Molecular Methods in Ionotropic Receptor Biology. Boca Raton (FL): CRC Press/Taylor & Francis; 2006. Chapter 5. In: Kittler JT, Moss SJ, editors. The Dynamic Synapse: Molecular Methods in Ionotropic Receptor Biology. Boca Raton (FL): CRC Press/Taylor & Francis; 2006. Chapter 5. PMID: 21204476 Free Books & Documents. Review.
Cited by
-
Mechanisms and functions of protein S-acylation.Nat Rev Mol Cell Biol. 2024 Jun;25(6):488-509. doi: 10.1038/s41580-024-00700-8. Epub 2024 Feb 14. Nat Rev Mol Cell Biol. 2024. PMID: 38355760 Review.
-
Ileal proteomic changes associated with IL-25-mediated resistance against intestinal trematode infections.Parasit Vectors. 2020 Jul 2;13(1):336. doi: 10.1186/s13071-020-04206-y. Parasit Vectors. 2020. PMID: 32616023 Free PMC article.
-
S-Palmitoylation of Synaptic Proteins in Neuronal Plasticity in Normal and Pathological Brains.Cells. 2023 Jan 21;12(3):387. doi: 10.3390/cells12030387. Cells. 2023. PMID: 36766729 Free PMC article. Review.
-
AP-4 regulates neuronal lysosome composition, function, and transport via regulating export of critical lysosome receptor proteins at the trans-Golgi network.Mol Biol Cell. 2022 Oct 1;33(12):ar102. doi: 10.1091/mbc.E21-09-0473. Epub 2022 Aug 17. Mol Biol Cell. 2022. PMID: 35976706 Free PMC article.
-
Endosomal Trafficking in Alzheimer's Disease, Parkinson's Disease, and Neuronal Ceroid Lipofuscinosis.Mol Cell Biol. 2020 Sep 14;40(19):e00262-20. doi: 10.1128/MCB.00262-20. Print 2020 Sep 14. Mol Cell Biol. 2020. PMID: 32690545 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources