

Host CARD11 Inhibits Newcastle Disease Virus Replication by Suppressing Viral Polymerase Activity in Neurons
- PMID: 31554683
- PMCID: PMC6880160
- DOI: 10.1128/JVI.01499-19
Host CARD11 Inhibits Newcastle Disease Virus Replication by Suppressing Viral Polymerase Activity in Neurons
Abstract
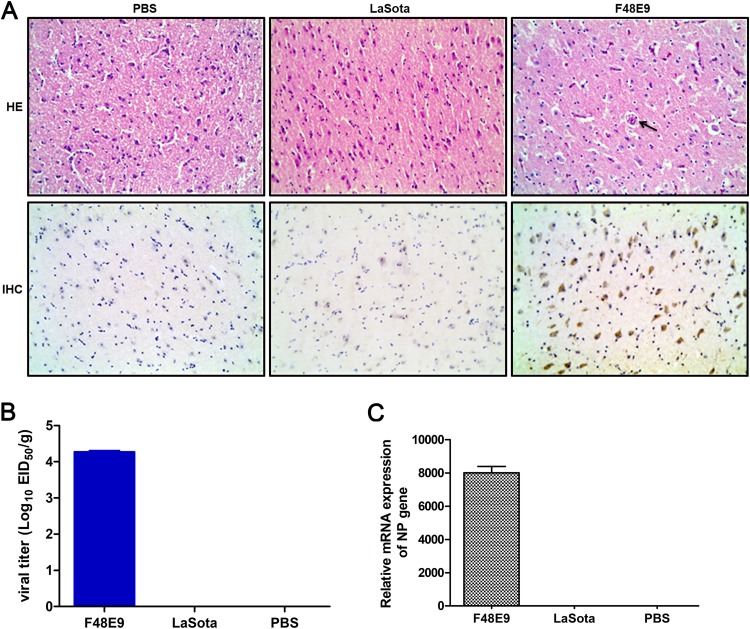
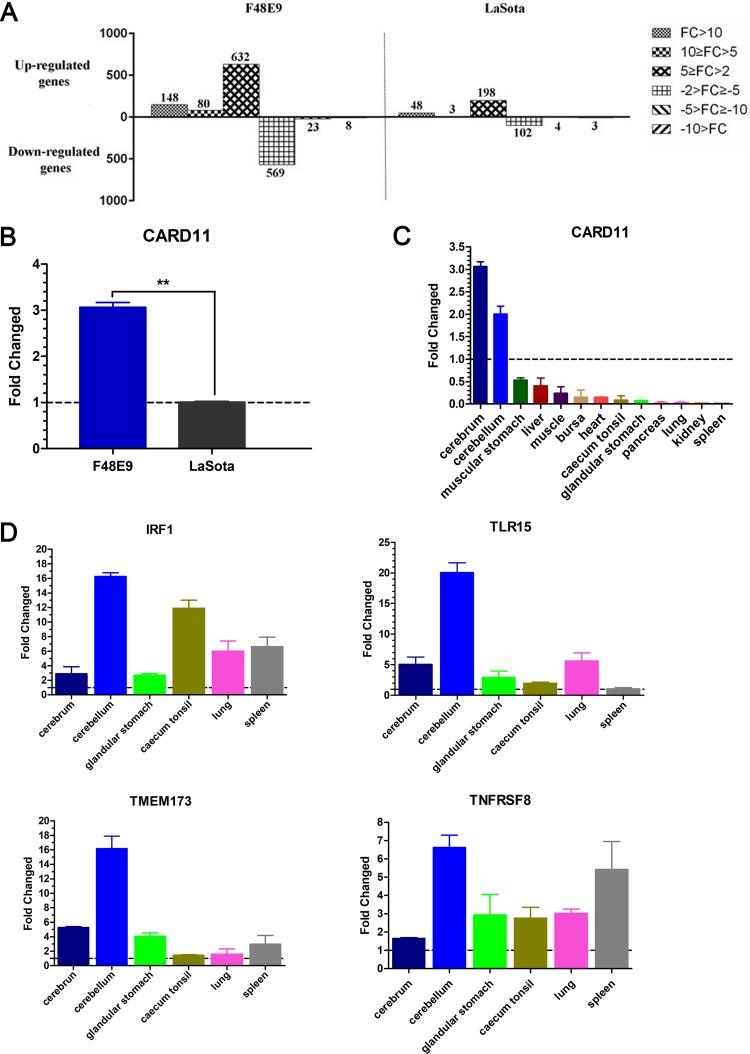
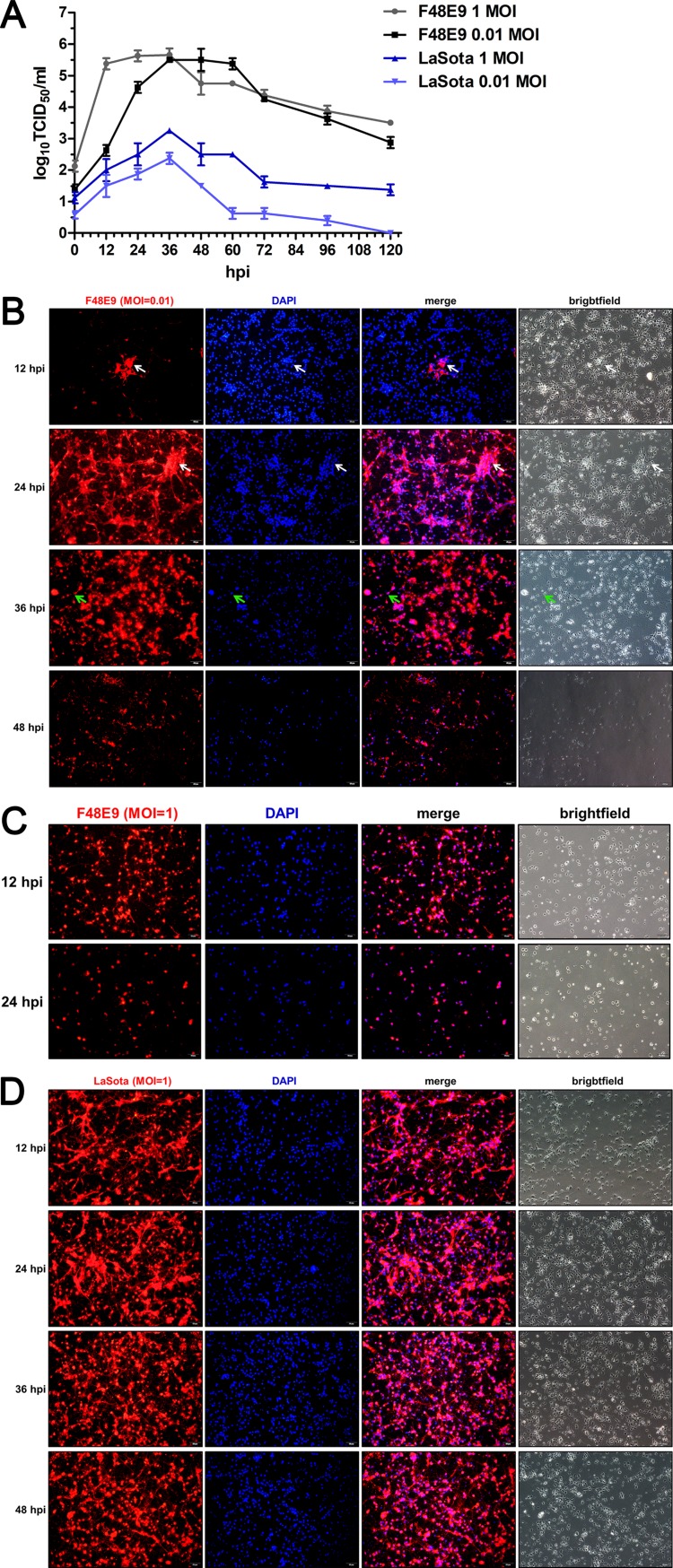
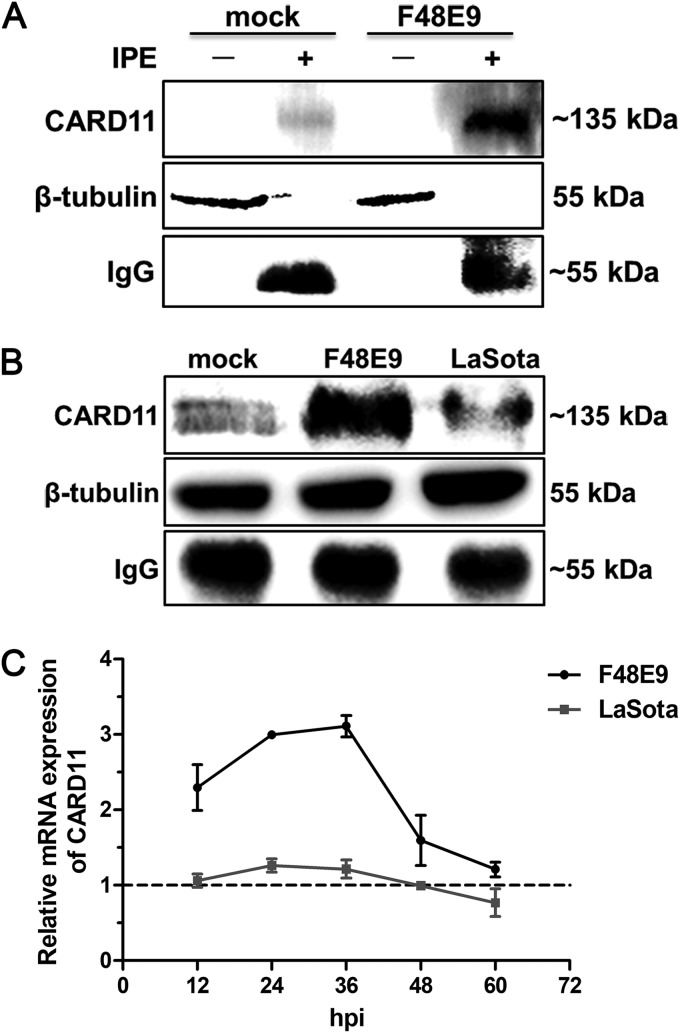
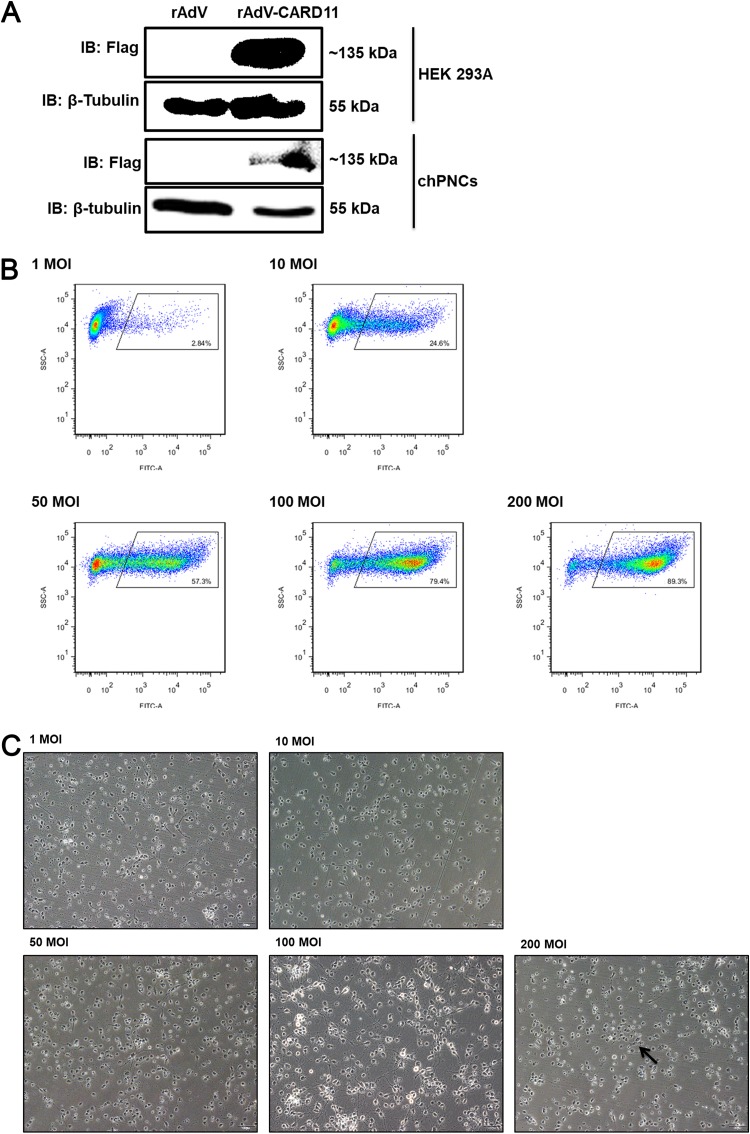
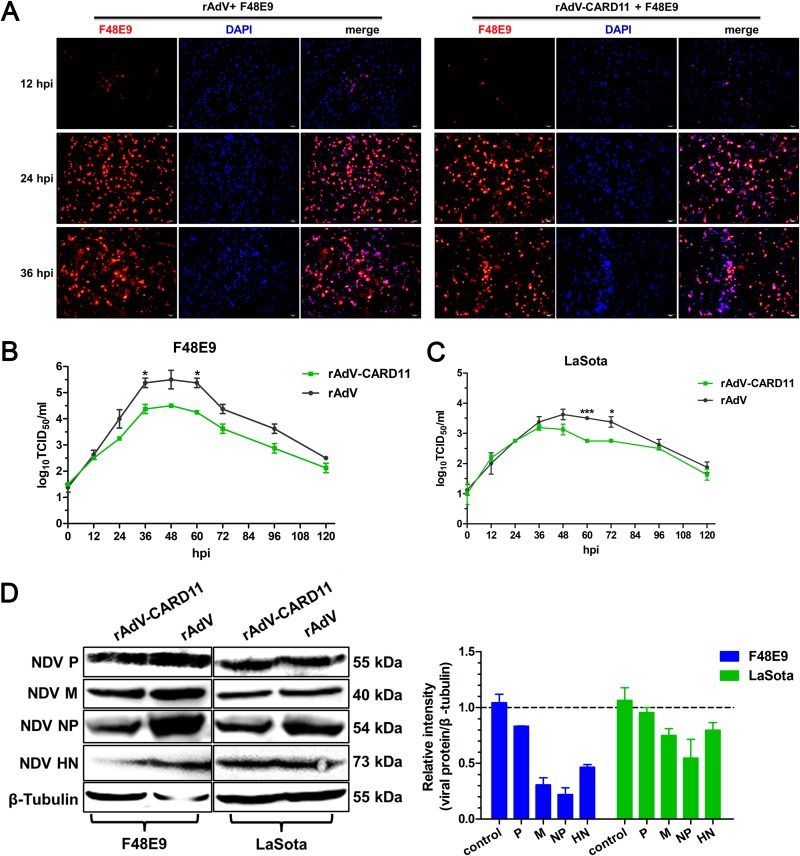
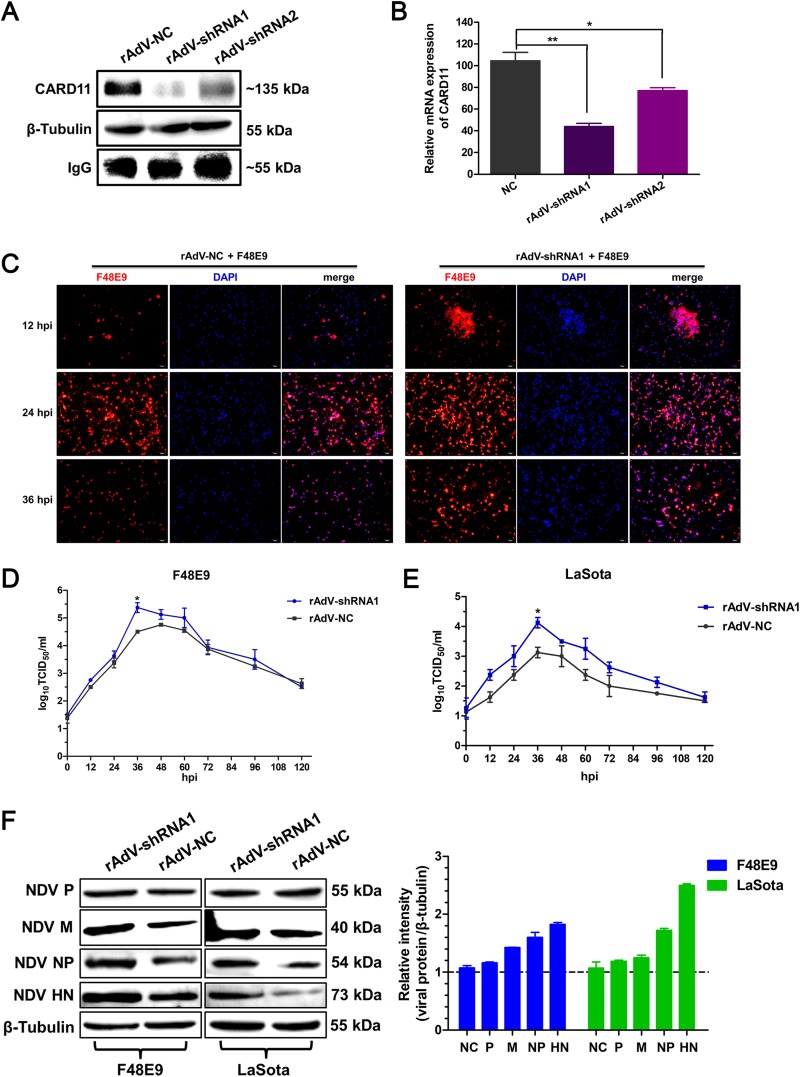
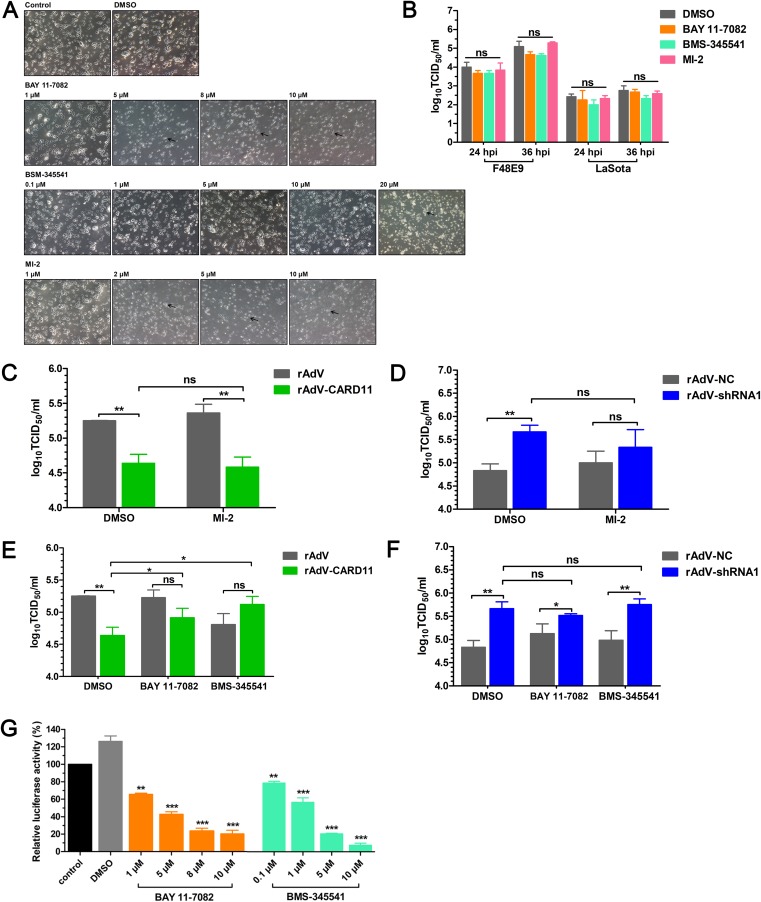
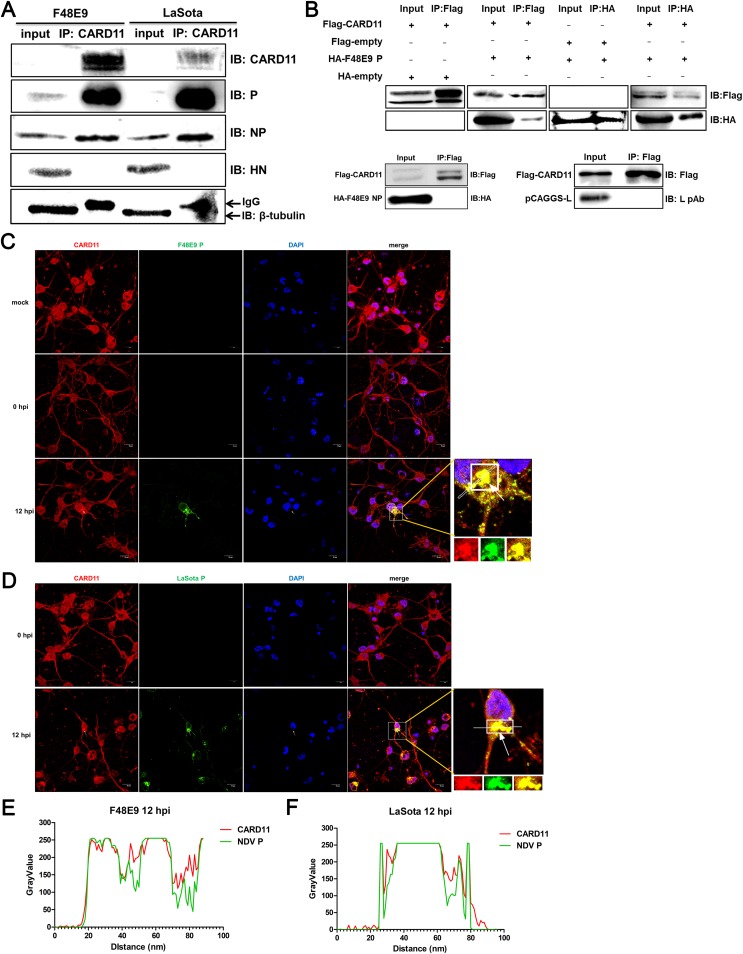
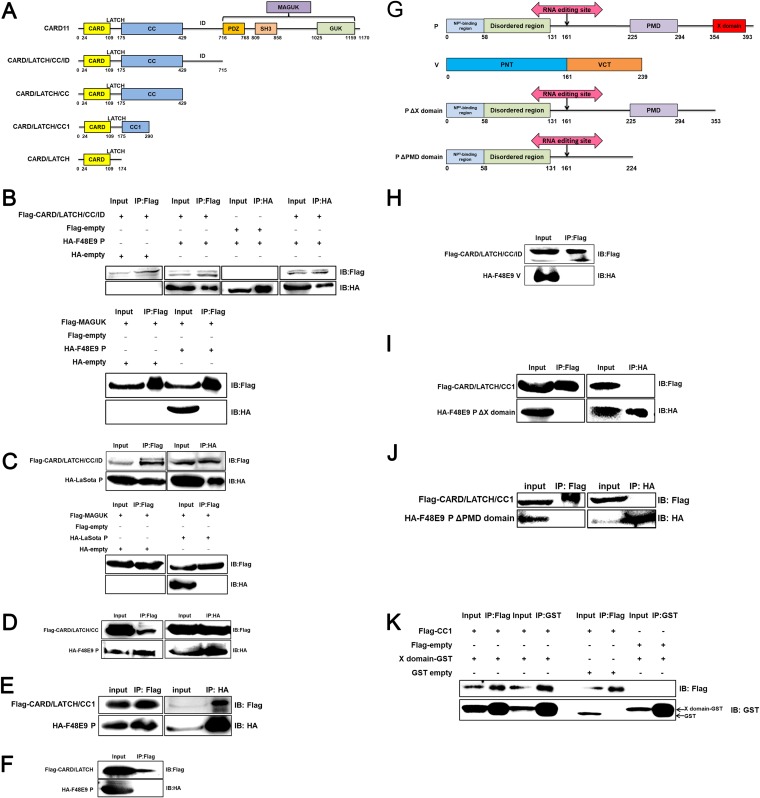
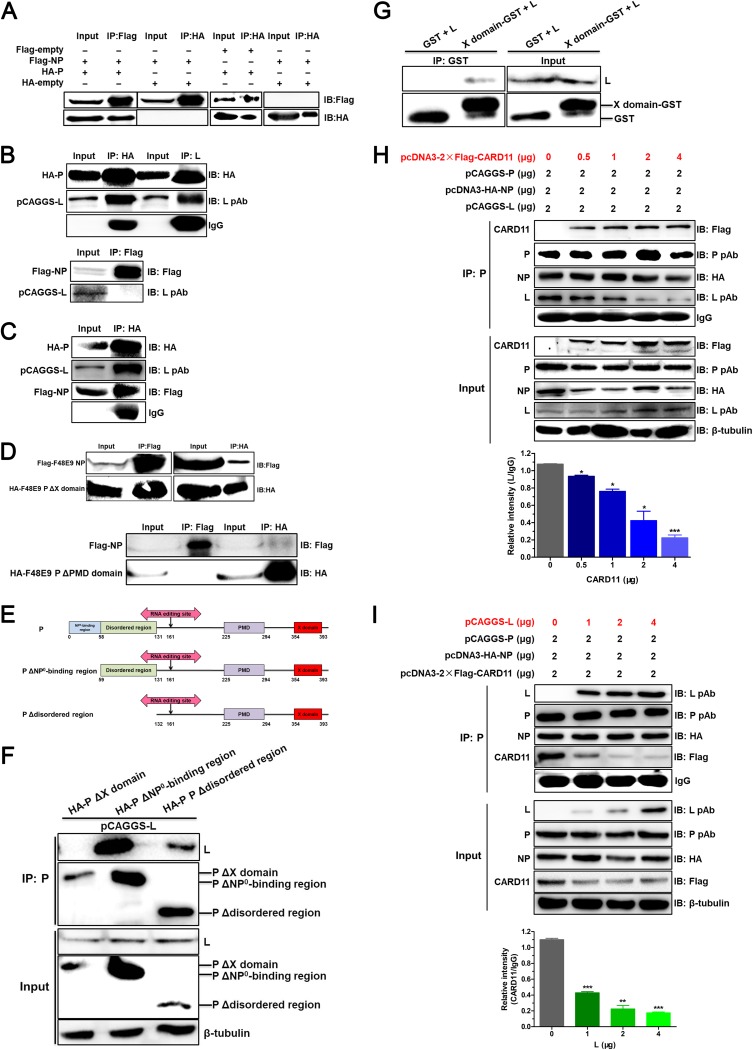
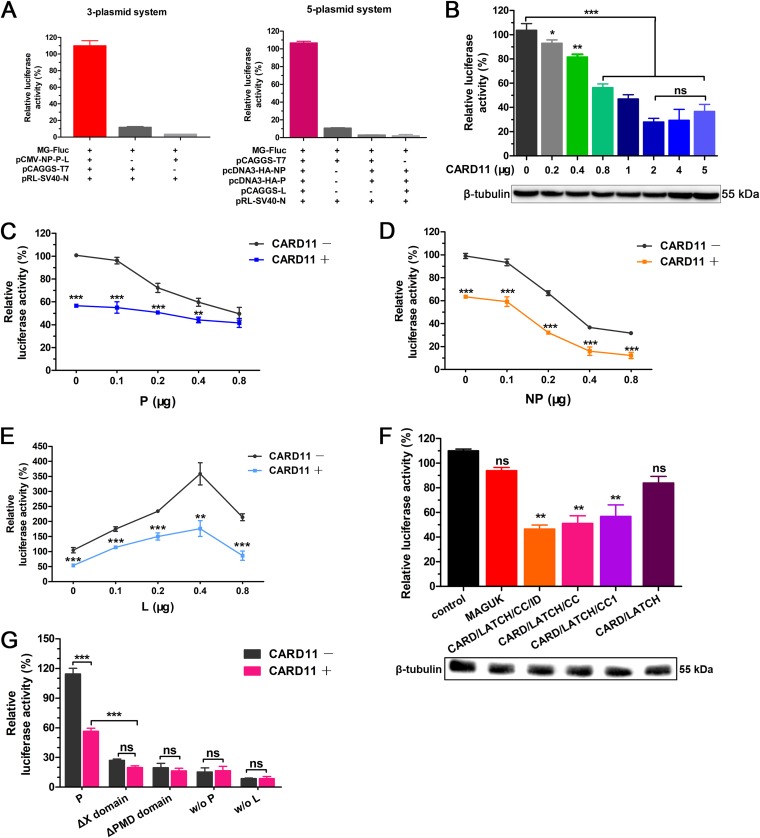
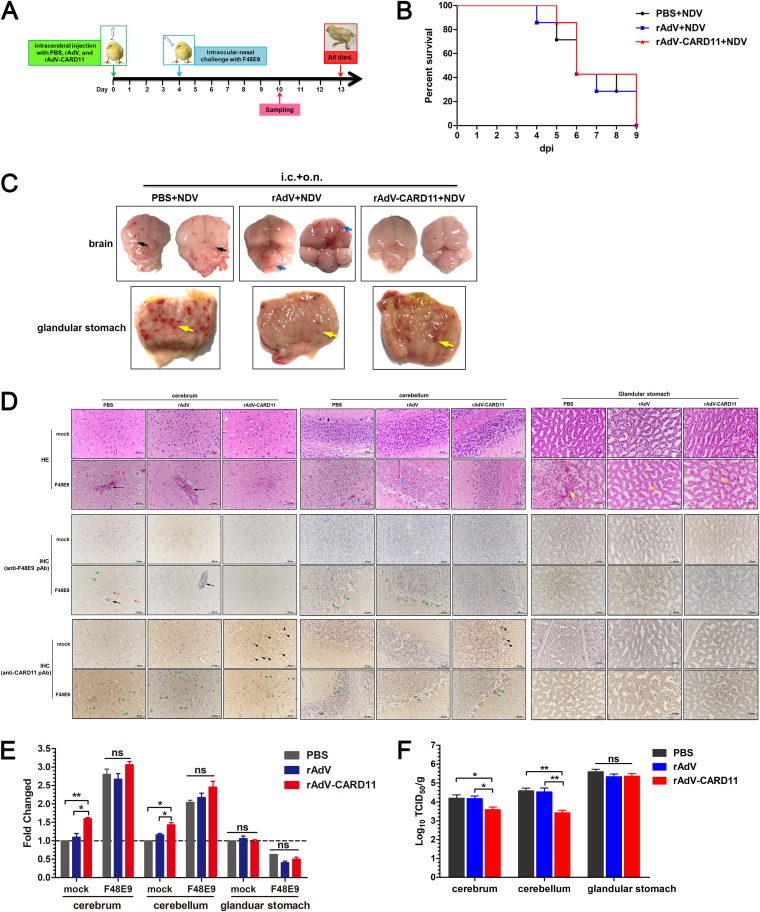
Host factors play multiple essential roles in the replication and pathogenesis of mammalian neurotropic viruses. However, the cellular proteins of the central nervous system (CNS) involved in avian neurotropic virus infection have not been completely elucidated. Here, we employed a gene microarray to identify caspase recruitment domain-containing protein 11 (CARD11), a lymphoma-associated scaffold protein presenting brain-specific upregulated expression in a virulent neurotropic Newcastle disease virus (NDV)-infected natural host. Chicken primary neuronal cells infected with NDV appeared slightly syncytial and died quickly. CARD11 overexpression inhibited viral replication and delayed cytopathic effects; conversely, depletion of CARD11 enhanced viral replication and cytopathic effects in chicken primary neuronal cells. The inhibition of viral replication by CARD11 could not be blocked with CARD11-Bcl10-MALT1 (CBM) signalosome and NF-κB signaling inhibitors. CARD11 was found to interact directly with the viral phosphoprotein (P) through its CC1 domain and the X domain of P; this X domain also mediated the interaction between P and the viral large polymerase protein (L). The CARD11 CC1 domain and L competitively bound to P via the X domain that hindered the P-L interaction of the viral ribonucleoprotein (RNP) complex, resulting in a reduction of viral polymerase activity in a minigenome assay and inhibition of viral replication. Animal experiments further revealed that CARD11 contributed to viral replication inhibition and neuropathology in infected chicken brains. Taken together, our findings identify CARD11 as a brain-specific antiviral factor of NDV infection in avian species.IMPORTANCE Newcastle disease virus (NDV) substantially impacts the poultry industry worldwide and causes viral encephalitis and neurological disorders leading to brain damage, paralysis, and death. The mechanism of interaction between this neurotropic virus and the avian central nervous system (CNS) is largely unknown. Here, we report that host protein CARD11 presented brain-specific upregulated expression that inhibited NDV replication, which was not due to CARD11-Bcl10-MALT1 (CBM) complex-triggered activation of its downstream signaling pathways. The inhibitory mechanism of viral replication is through the CARD11 CC1 domain, and the viral large polymerase protein (L) competitively interacts with the X domain of the viral phosphoprotein (P), which hampers the P-L interaction, suppressing the viral polymerase activity and viral replication. An in vivo study indicated that CARD11 alleviated neuropathological lesions and reduced viral replication in chicken brains. These results provide insight into the interaction between NDV infection and the host defense in the CNS and a potential antiviral target for viral neural diseases.
Keywords: CARD11; Newcastle disease virus; avian neurons; brain-specific upregulation; competitive binding; inhibition to viral replication; viral RNP; viral polymerase activity.
Copyright © 2019 American Society for Microbiology.
Figures
Similar articles
-
Cellular CARD11 Inhibits the Fusogenic Activity of Newcastle Disease Virus via CBM Signalosome-Mediated Furin Reduction in Chicken Fibroblasts.Front Microbiol. 2021 Feb 2;12:607451. doi: 10.3389/fmicb.2021.607451. eCollection 2021. Front Microbiol. 2021. PMID: 33603723 Free PMC article.
-
Molecular Determinants of Scaffold-induced Linear Ubiquitinylation of B Cell Lymphoma/Leukemia 10 (Bcl10) during T Cell Receptor and Oncogenic Caspase Recruitment Domain-containing Protein 11 (CARD11) Signaling.J Biol Chem. 2016 Dec 9;291(50):25921-25936. doi: 10.1074/jbc.M116.754028. Epub 2016 Oct 24. J Biol Chem. 2016. PMID: 27777308 Free PMC article.
-
Lymphomagenic CARD11/BCL10/MALT1 signaling drives malignant B-cell proliferation via cooperative NF-κB and JNK activation.Proc Natl Acad Sci U S A. 2015 Dec 29;112(52):E7230-8. doi: 10.1073/pnas.1507459112. Epub 2015 Dec 14. Proc Natl Acad Sci U S A. 2015. PMID: 26668357 Free PMC article.
-
The CBM-opathies-A Rapidly Expanding Spectrum of Human Inborn Errors of Immunity Caused by Mutations in the CARD11-BCL10-MALT1 Complex.Front Immunol. 2018 Sep 19;9:2078. doi: 10.3389/fimmu.2018.02078. eCollection 2018. Front Immunol. 2018. PMID: 30283440 Free PMC article. Review.
-
Genetic errors of the human caspase recruitment domain-B-cell lymphoma 10-mucosa-associated lymphoid tissue lymphoma-translocation gene 1 (CBM) complex: Molecular, immunologic, and clinical heterogeneity.J Allergy Clin Immunol. 2015 Nov;136(5):1139-49. doi: 10.1016/j.jaci.2015.06.031. Epub 2015 Aug 12. J Allergy Clin Immunol. 2015. PMID: 26277595 Free PMC article. Review.
Cited by
-
The Dual Roles of Activating Transcription Factor 3 (ATF3) in Inflammation, Apoptosis, Ferroptosis, and Pathogen Infection Responses.Int J Mol Sci. 2024 Jan 9;25(2):824. doi: 10.3390/ijms25020824. Int J Mol Sci. 2024. PMID: 38255898 Free PMC article. Review.
-
Cellular CARD11 Inhibits the Fusogenic Activity of Newcastle Disease Virus via CBM Signalosome-Mediated Furin Reduction in Chicken Fibroblasts.Front Microbiol. 2021 Feb 2;12:607451. doi: 10.3389/fmicb.2021.607451. eCollection 2021. Front Microbiol. 2021. PMID: 33603723 Free PMC article.
-
First Isolation and Characteristics of Bovine Parainfluenza Virus Type 3 from Yaks.Pathogens. 2022 Aug 24;11(9):962. doi: 10.3390/pathogens11090962. Pathogens. 2022. PMID: 36145395 Free PMC article.
-
Isolation and Pathogenic Characterization of Pigeon Paramyxovirus Type 1 via Different Inoculation Routes in Pigeons.Front Vet Sci. 2021 Feb 17;7:569901. doi: 10.3389/fvets.2020.569901. eCollection 2020. Front Vet Sci. 2021. PMID: 33681314 Free PMC article.
-
The multifaceted interactions between Newcastle disease virus proteins and host proteins: a systematic review.Virulence. 2024 Dec;15(1):2299182. doi: 10.1080/21505594.2023.2299182. Epub 2024 Jan 9. Virulence. 2024. PMID: 38193514 Free PMC article.
References
-
- Ludlow M, Kortekaas J, Herden C, Hoffmann B, Tappe D, Trebst C, Griffin DE, Brindle HE, Solomon T, Brown AS, van Riel D, Wolthers KC, Pajkrt D, Wohlsein P, Martina BEE, Baumgartner W, Verjans GM, Osterhaus A. 2016. Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol 131:159–184. doi:10.1007/s00401-015-1511-3. - DOI - PMC - PubMed
-
- Chen Z, Wang X, Ashraf U, Zheng B, Ye J, Zhou D, Zhang H, Song Y, Chen H, Zhao S, Cao S. 2018. Activation of neuronal N-methyl-d-aspartate receptor plays a pivotal role in Japanese encephalitis virus-induced neuronal cell damage. J Neuroinflammation 15:238. doi:10.1186/s12974-018-1280-8. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
