

Extensive tests and evaluation of the CHARMM36IDPSFF force field for intrinsically disordered proteins and folded proteins
- PMID: 31552948
- PMCID: PMC7198049
- DOI: 10.1039/c9cp03434j
Extensive tests and evaluation of the CHARMM36IDPSFF force field for intrinsically disordered proteins and folded proteins
Abstract
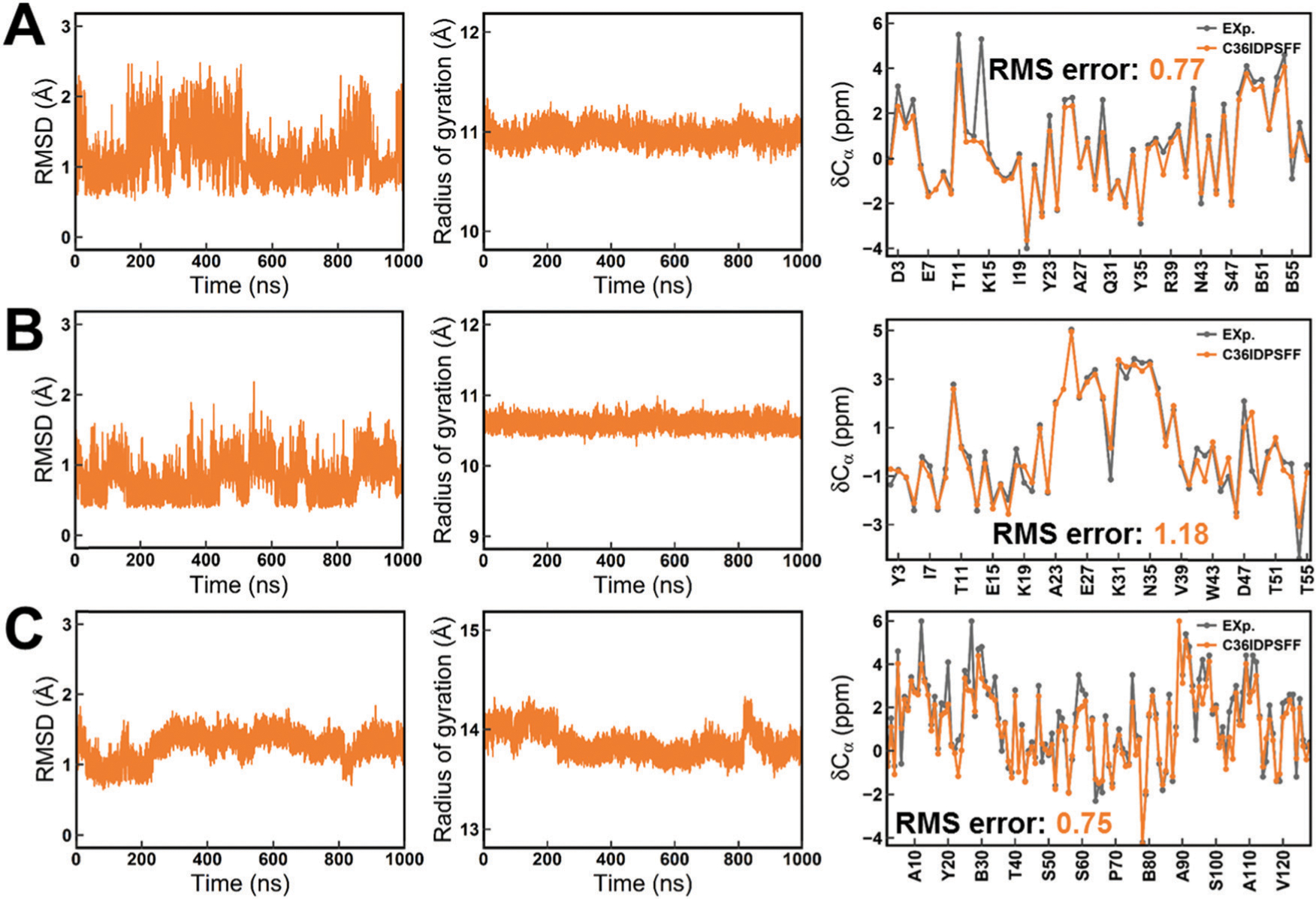
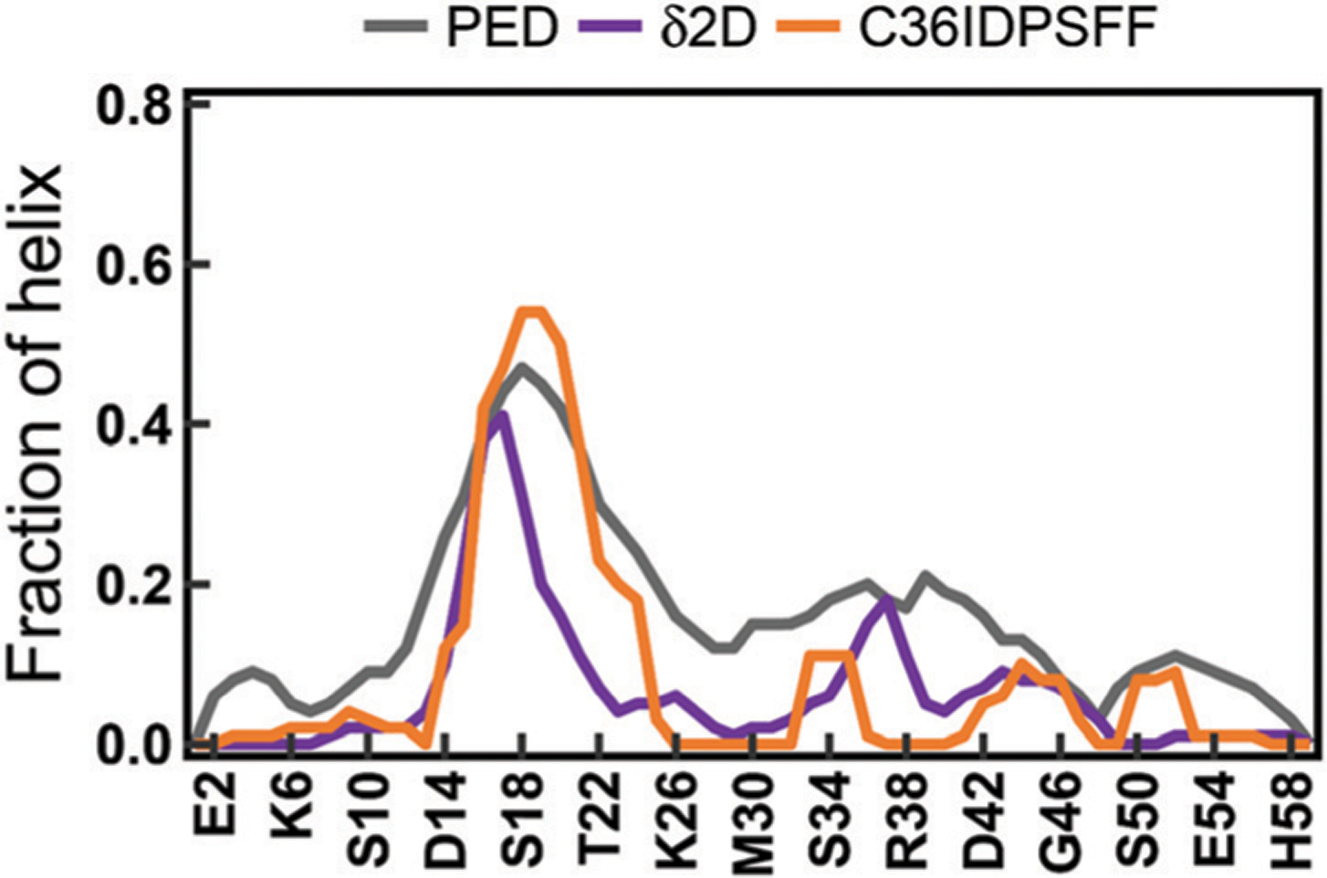
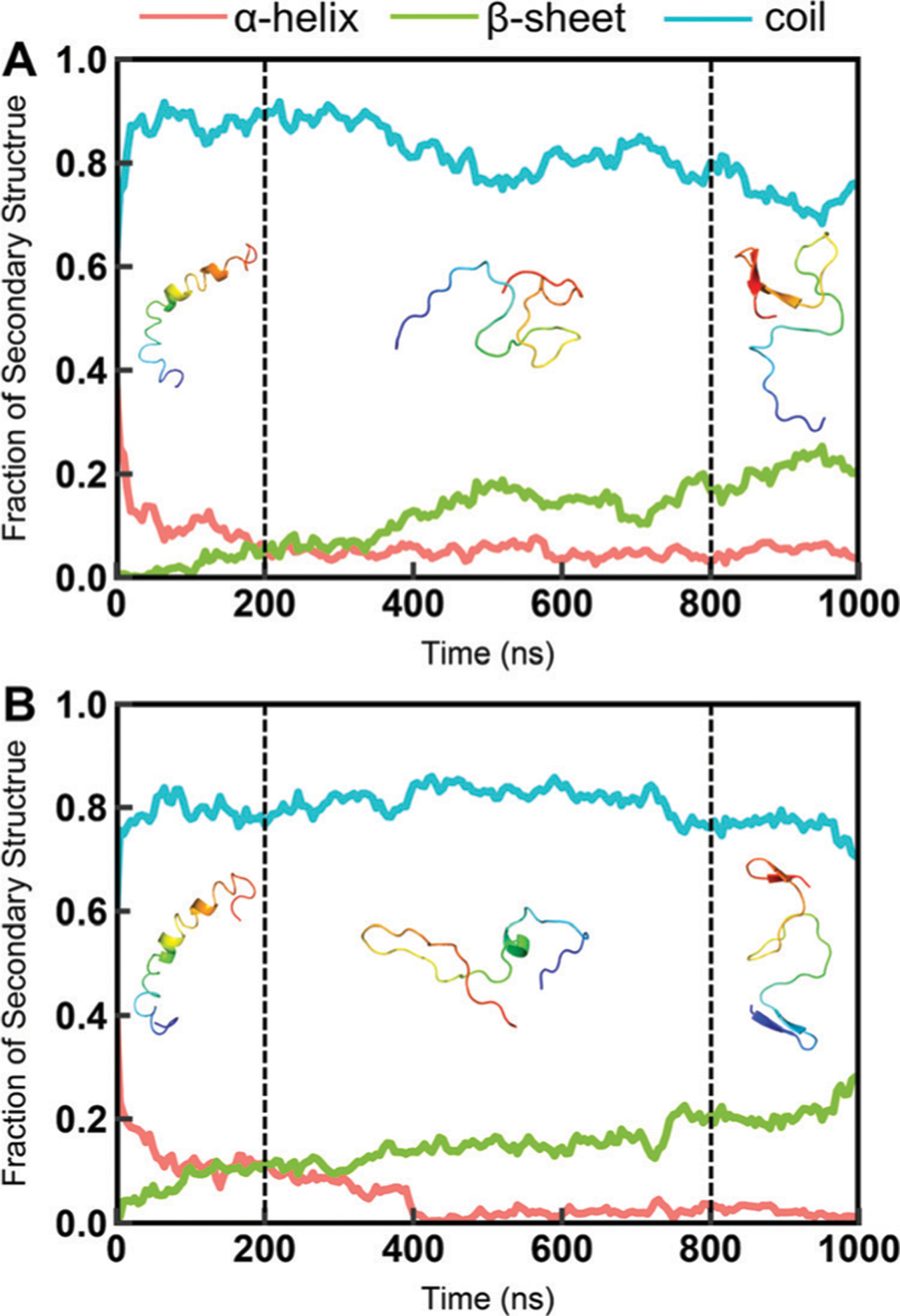
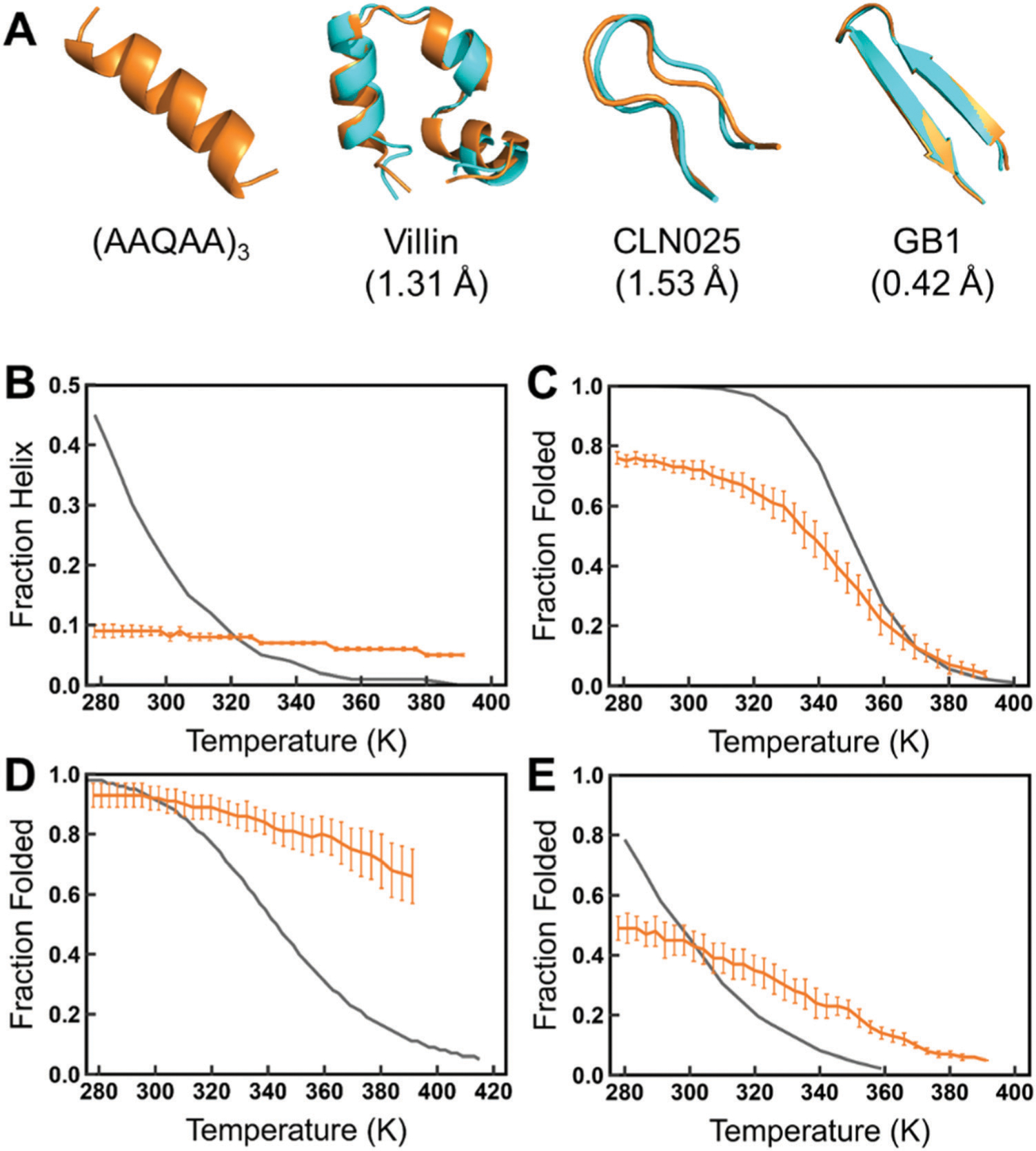
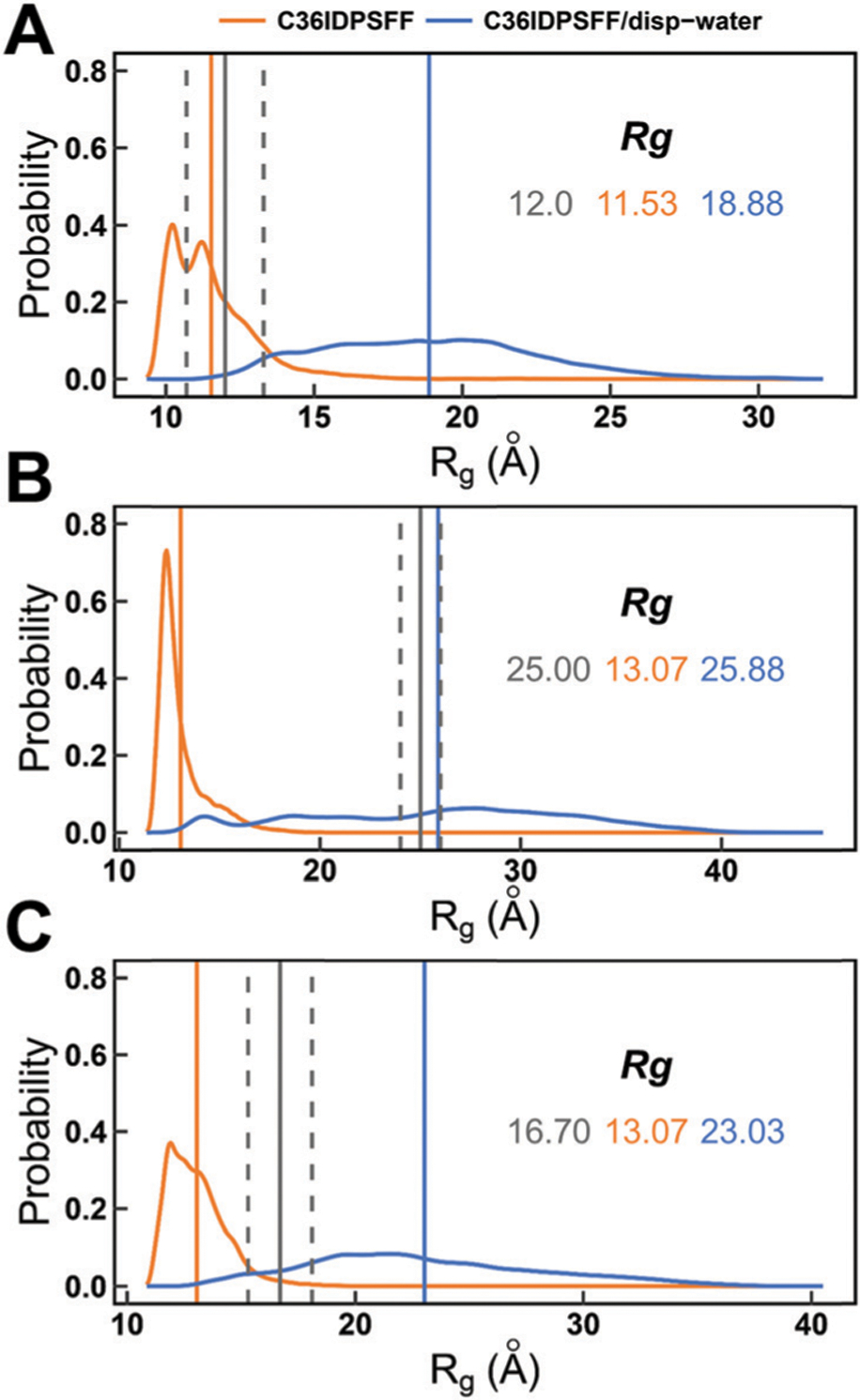
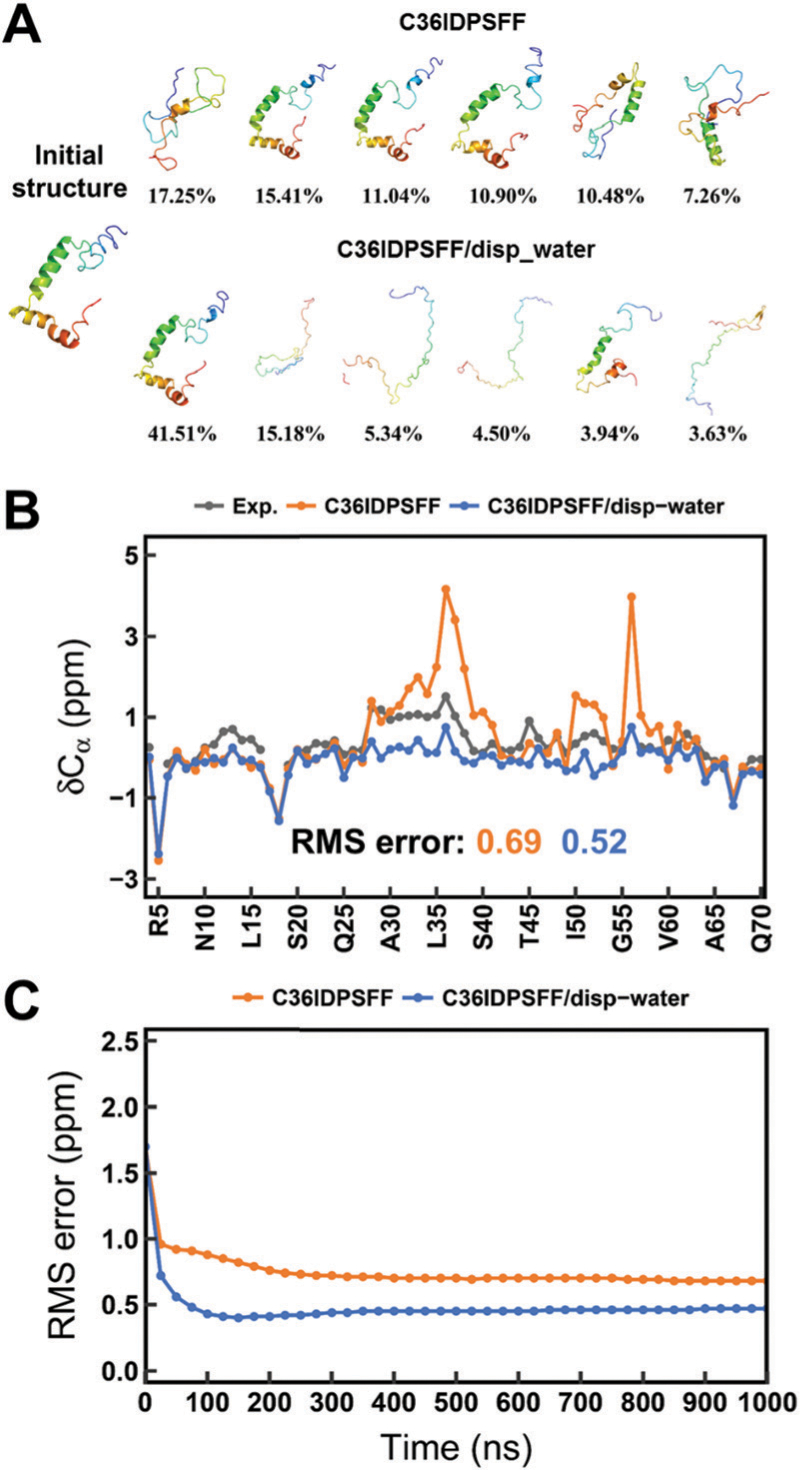
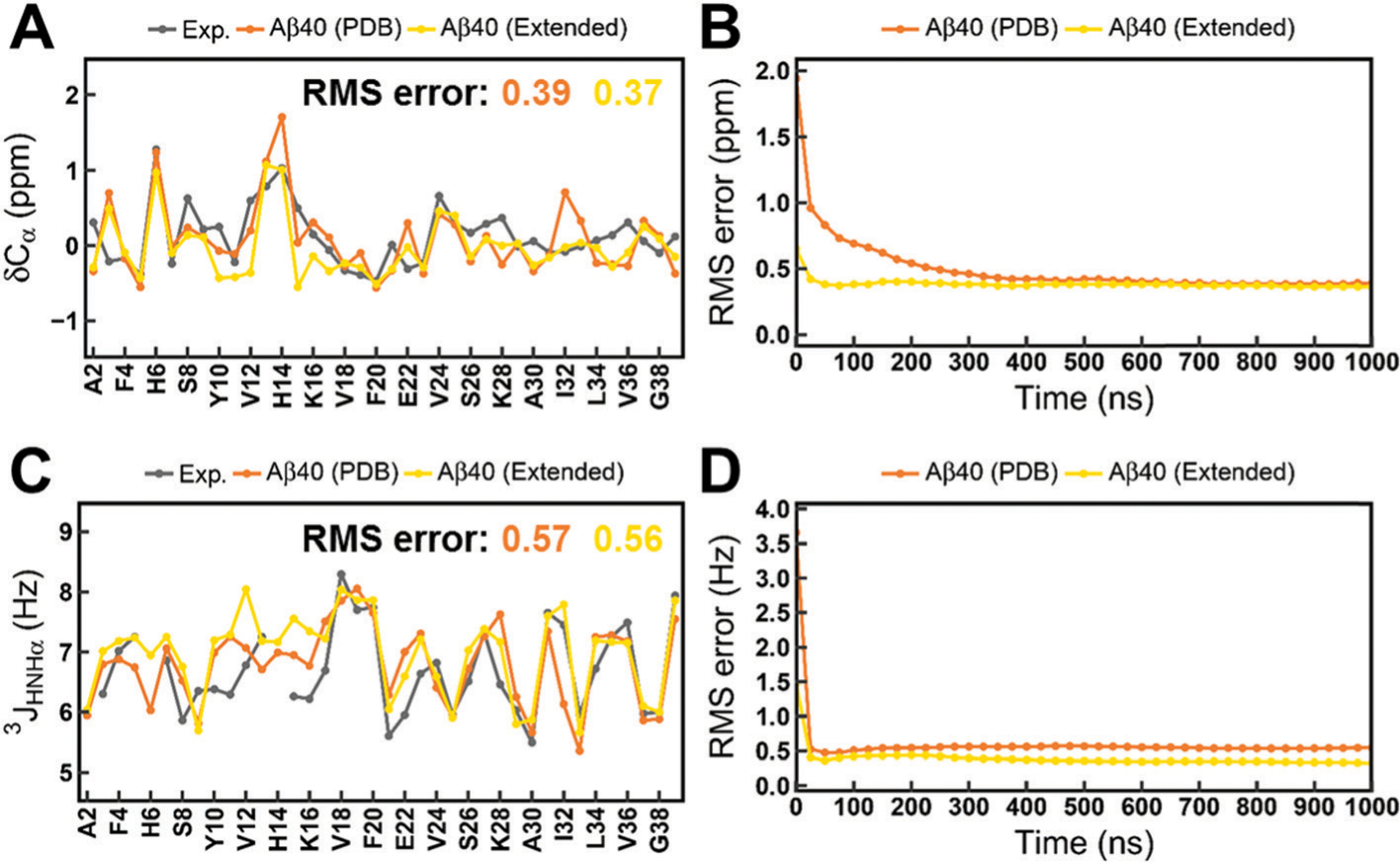
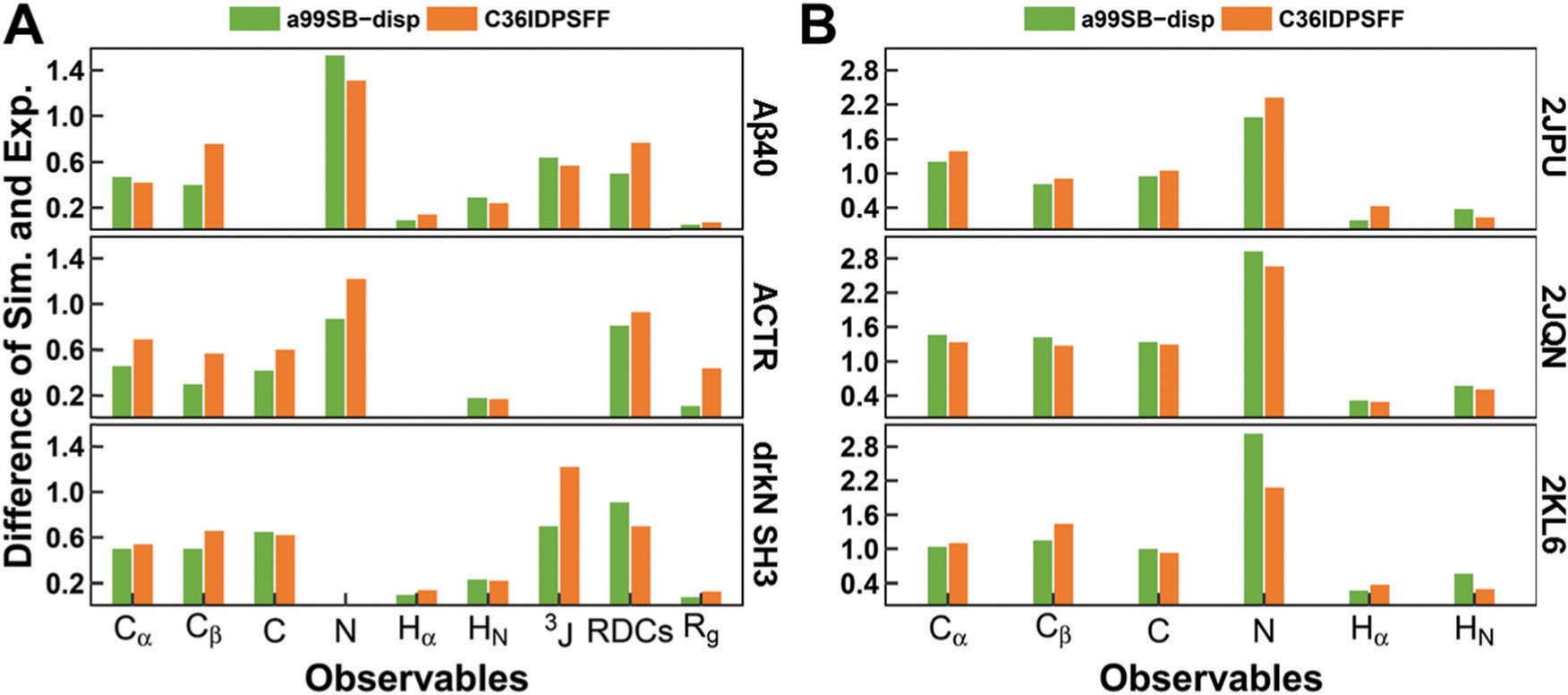
Intrinsically disordered proteins (IDPs) have received increasing attention in recent studies due to their structural heterogeneity and critical biological functions. To fully understand the structural properties and determine accurate ensembles of IDPs, molecular dynamics (MD) simulation was widely used to sample diverse conformations and reveal the structural dynamics. However, the classical state-of-the-art force fields perform well for folded proteins while being unsatisfactory for the simulations of disordered proteins reported in many previous studies. Thus, improved force fields were developed to precisely describe both folded proteins and disordered proteins. Preliminary tests show that our newly developed CHARMM36IDPSFF (C36IDPSFF) force field can well reproduce the experimental observables of several disordered proteins, but more tests on different types of proteins are needed to further evaluate the performance of C36IDPSFF. Here, we extensively simulate short peptides, disordered proteins, and fast-folding proteins as well as folded proteins, and compare the simulated results with the experimental observables. The simulation results show that C36IDPSFF could substantially reproduce the experimental observables for most of the tested proteins but some limitations are also found in the radius of gyration of large disordered proteins and the stability of fast-folding proteins. This force field will facilitate large scale studies of protein structural dynamics and functions using MD simulations.
Conflict of interest statement
Conflicts of interest
The authors declare that there is no conflict of interest.
Figures
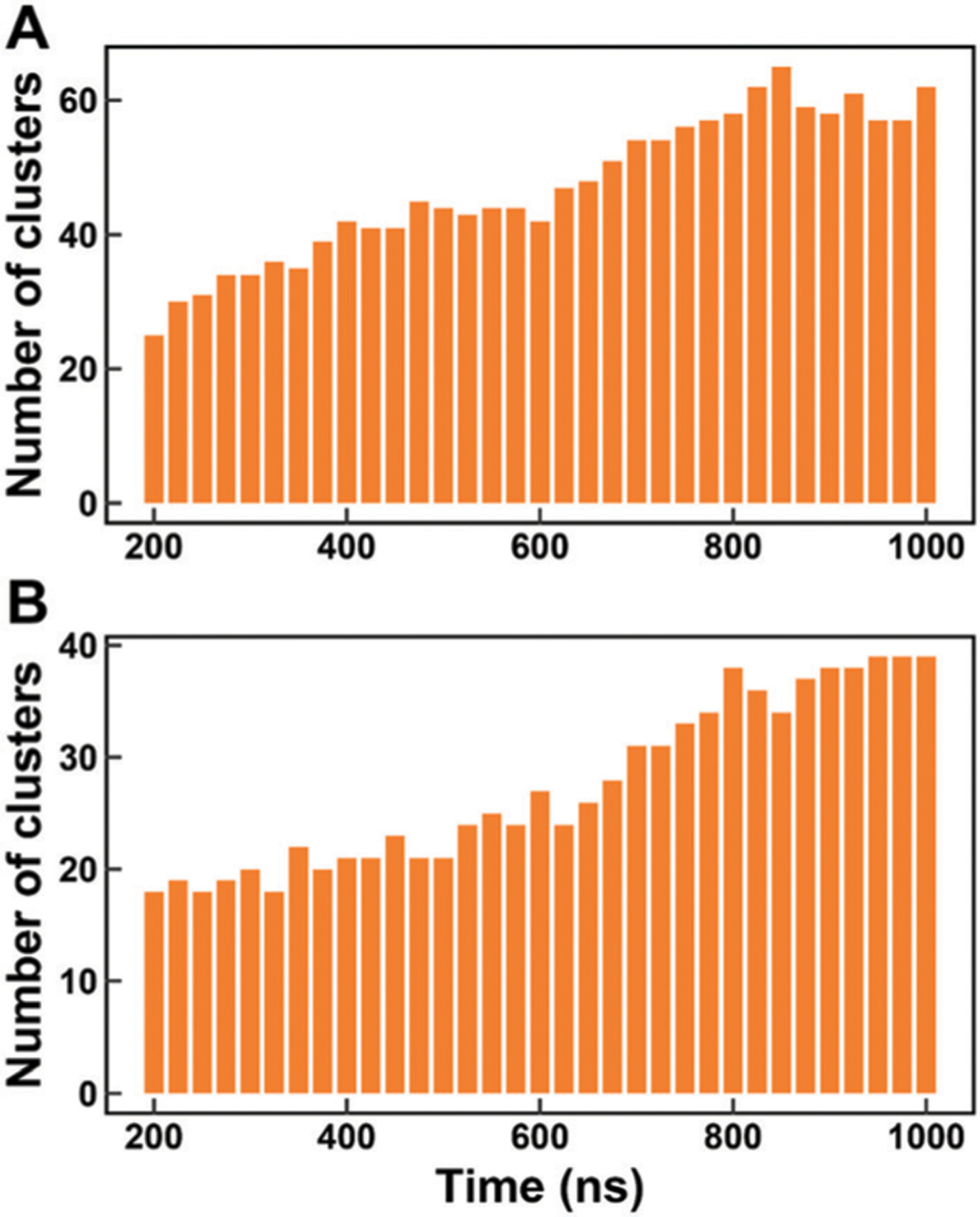
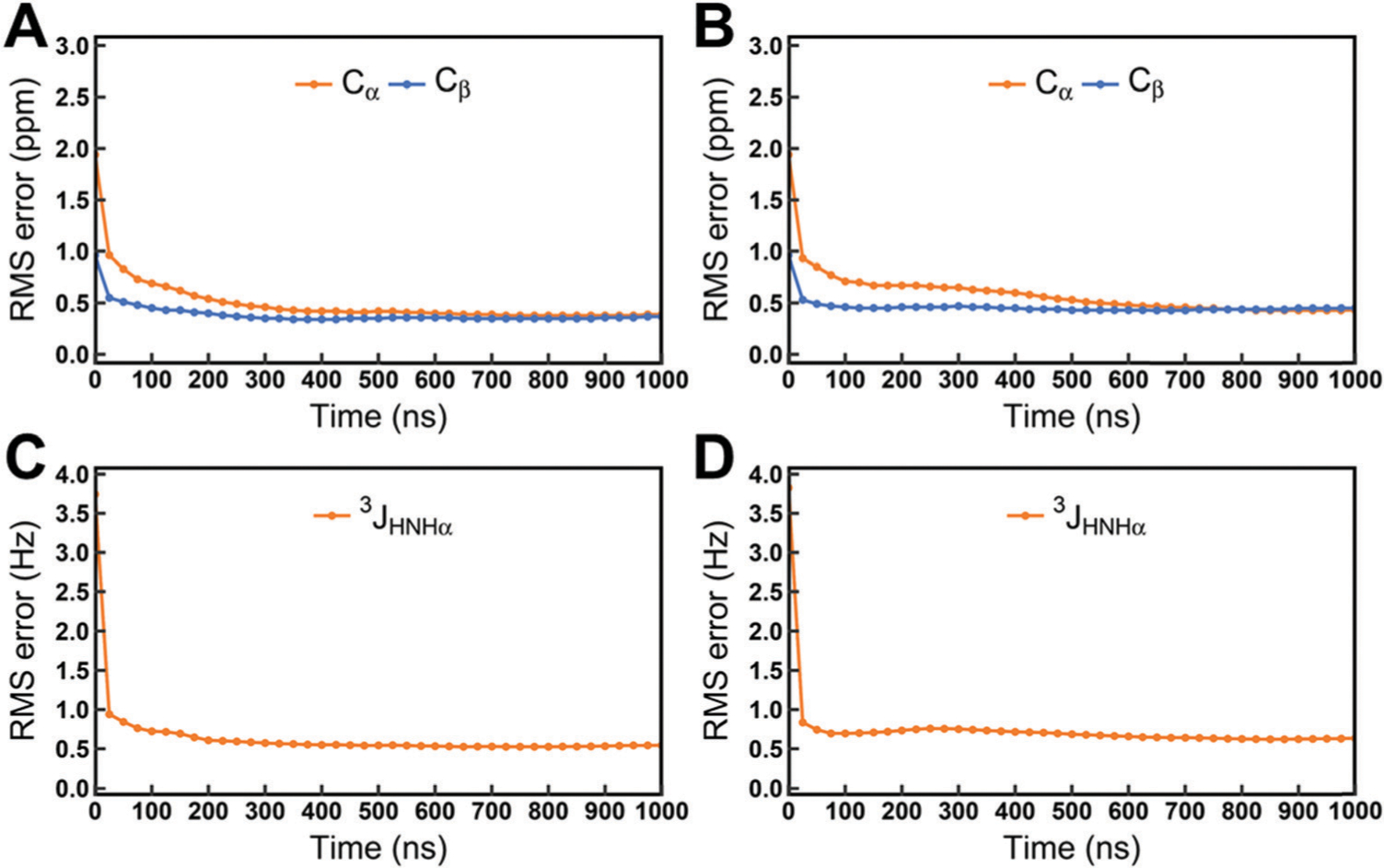
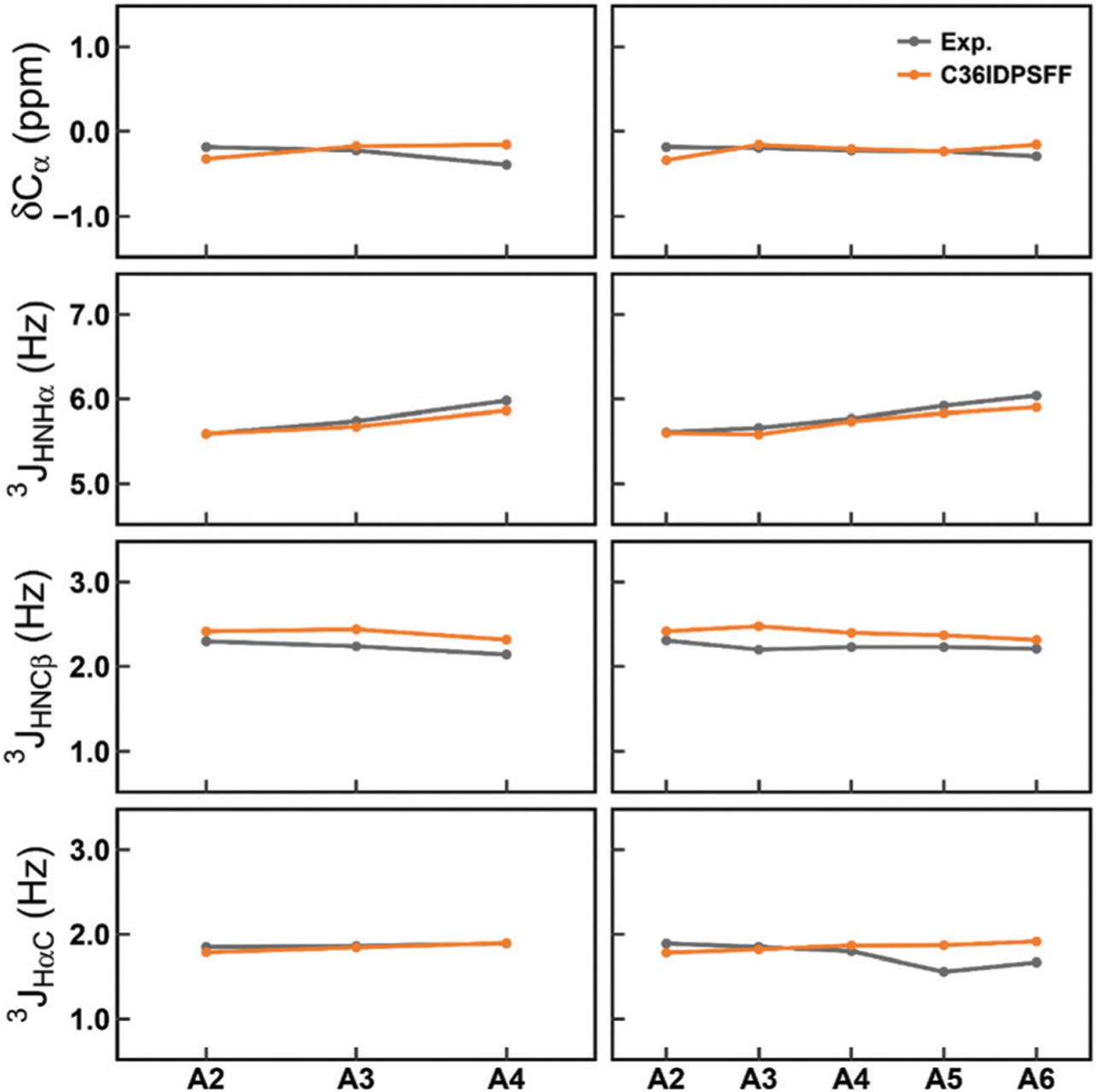
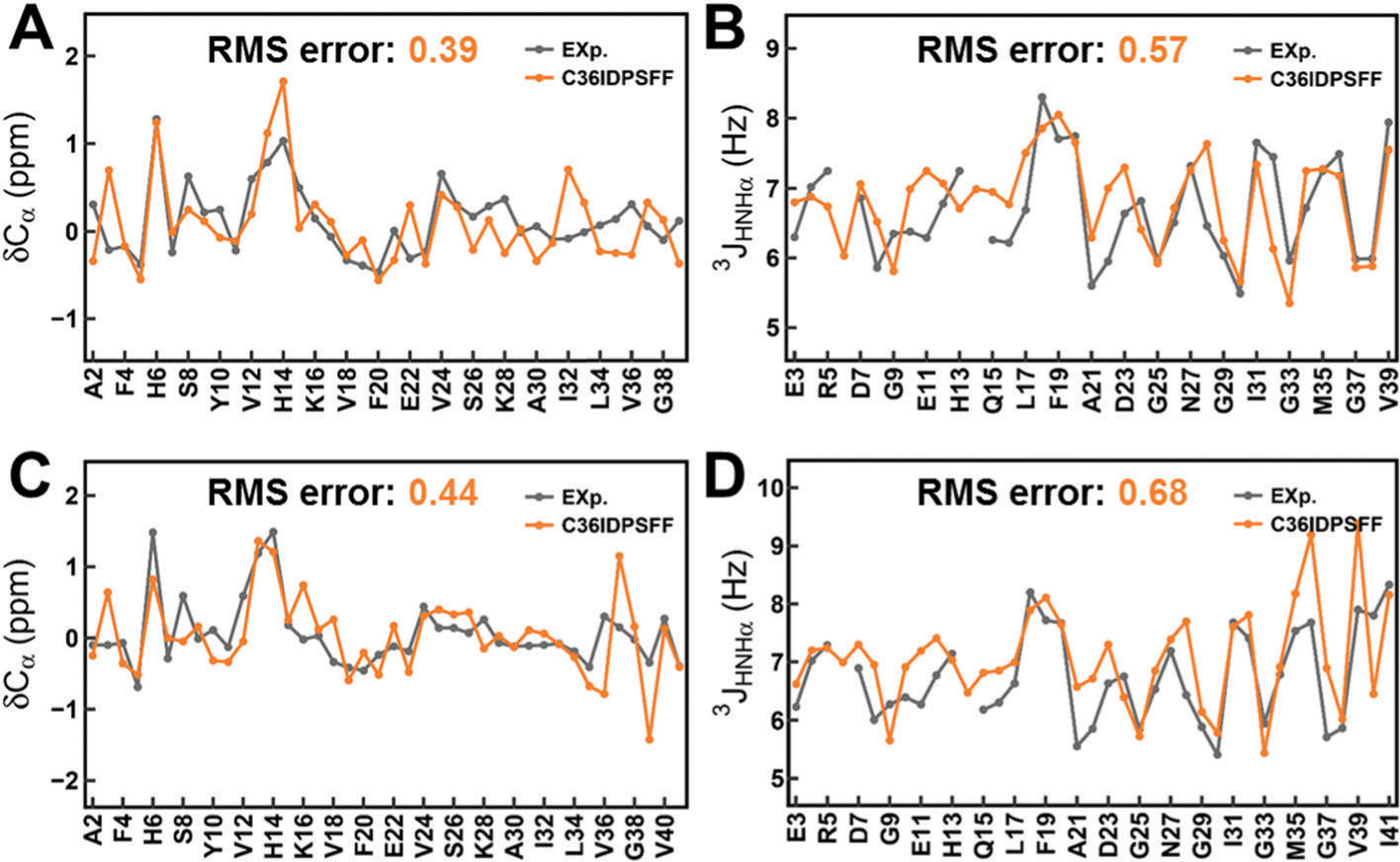
Similar articles
-
Residue-Specific Force Field Improving the Sample of Intrinsically Disordered Proteins and Folded Proteins.J Chem Inf Model. 2019 Nov 25;59(11):4793-4805. doi: 10.1021/acs.jcim.9b00647. Epub 2019 Oct 22. J Chem Inf Model. 2019. PMID: 31613621
-
Intrinsically disordered protein-specific force field CHARMM36IDPSFF.Chem Biol Drug Des. 2018 Oct;92(4):1722-1735. doi: 10.1111/cbdd.13342. Epub 2018 Jul 1. Chem Biol Drug Des. 2018. PMID: 29808548
-
Developing a molecular dynamics force field for both folded and disordered protein states.Proc Natl Acad Sci U S A. 2018 May 22;115(21):E4758-E4766. doi: 10.1073/pnas.1800690115. Epub 2018 May 7. Proc Natl Acad Sci U S A. 2018. PMID: 29735687 Free PMC article.
-
Molecular Dynamics Simulations Combined with Nuclear Magnetic Resonance and/or Small-Angle X-ray Scattering Data for Characterizing Intrinsically Disordered Protein Conformational Ensembles.J Chem Inf Model. 2019 May 28;59(5):1743-1758. doi: 10.1021/acs.jcim.8b00928. Epub 2019 Mar 18. J Chem Inf Model. 2019. PMID: 30840442 Review.
-
Recent advances in atomic molecular dynamics simulation of intrinsically disordered proteins.Phys Chem Chem Phys. 2021 Jan 21;23(2):777-784. doi: 10.1039/d0cp05818a. Phys Chem Chem Phys. 2021. PMID: 33355572 Review.
Cited by
-
Force Field Effects in Simulations of Flexible Peptides with Varying Polyproline II Propensity.J Chem Theory Comput. 2021 Oct 12;17(10):6634-6646. doi: 10.1021/acs.jctc.1c00408. Epub 2021 Sep 15. J Chem Theory Comput. 2021. PMID: 34524800 Free PMC article.
-
Recent Force Field Strategies for Intrinsically Disordered Proteins.J Chem Inf Model. 2021 Mar 22;61(3):1037-1047. doi: 10.1021/acs.jcim.0c01175. Epub 2021 Feb 16. J Chem Inf Model. 2021. PMID: 33591749 Free PMC article. Review.
-
High resolution ensemble description of metamorphic and intrinsically disordered proteins using an efficient hybrid parallel tempering scheme.Nat Commun. 2021 Feb 11;12(1):958. doi: 10.1038/s41467-021-21105-7. Nat Commun. 2021. PMID: 33574233 Free PMC article.
-
Fisetin inhibits tau aggregation by interacting with the protein and preventing the formation of β-strands.Int J Biol Macromol. 2021 May 1;178:381-393. doi: 10.1016/j.ijbiomac.2021.02.210. Epub 2021 Mar 1. Int J Biol Macromol. 2021. PMID: 33662414 Free PMC article.
-
Computational Models for the Study of Protein Aggregation.Methods Mol Biol. 2022;2340:51-78. doi: 10.1007/978-1-0716-1546-1_4. Methods Mol Biol. 2022. PMID: 35167070
References
-
- Ozenne V, Schneider R, Yao MX, Huang JR, Salmon L, Zweckstetter M, Jensen MR and Blackledge M, Mapping the Potential Energy Landscape of Intrinsically Disordered Proteins at Amino Acid Resolution, J. Am. Chem. Soc, 2012, 134, 15138–15148. - PubMed
-
- Dyson HJ and Wright PE, Intrinsically unstructured proteins and their functions, Nat. Rev. Mol. Cell Biol, 2005, 6, 197–208. - PubMed
-
- Skrabana R, Skrabanova-Khuebachova M, Kontsek P and Novak M, Alzheimer’s-disease-associated conformation of intrinsically disordered tau protein studied by intrinsically disordered protein liquid-phase competitive enzyme-linked immunosorbent assay, Anal. Biochem, 2006, 359, 230–237. - PubMed