

Targeting Autophagy for Overcoming Resistance to Anti-EGFR Treatments
- PMID: 31527477
- PMCID: PMC6769649
- DOI: 10.3390/cancers11091374
Targeting Autophagy for Overcoming Resistance to Anti-EGFR Treatments
Abstract
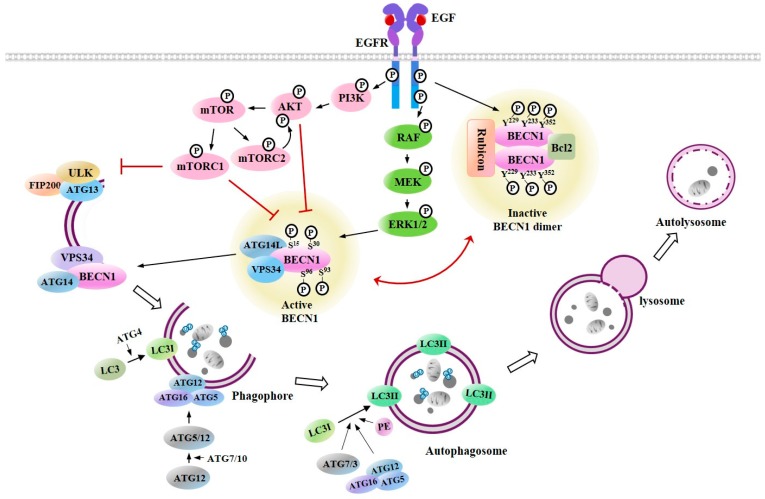
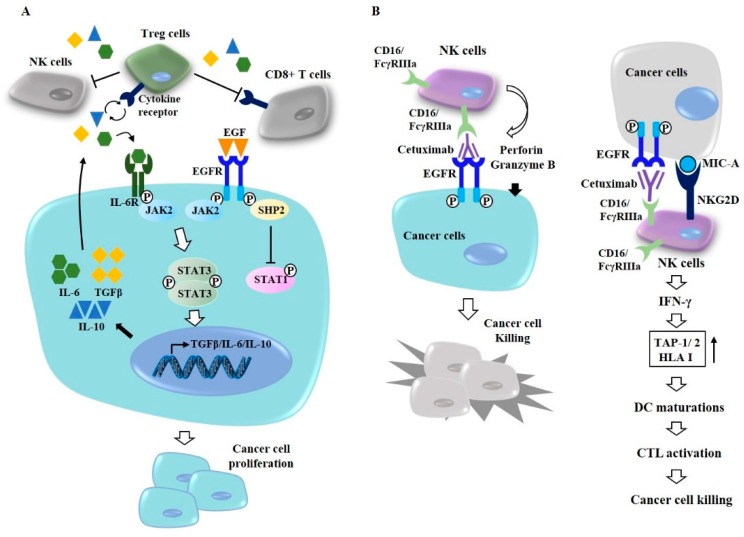
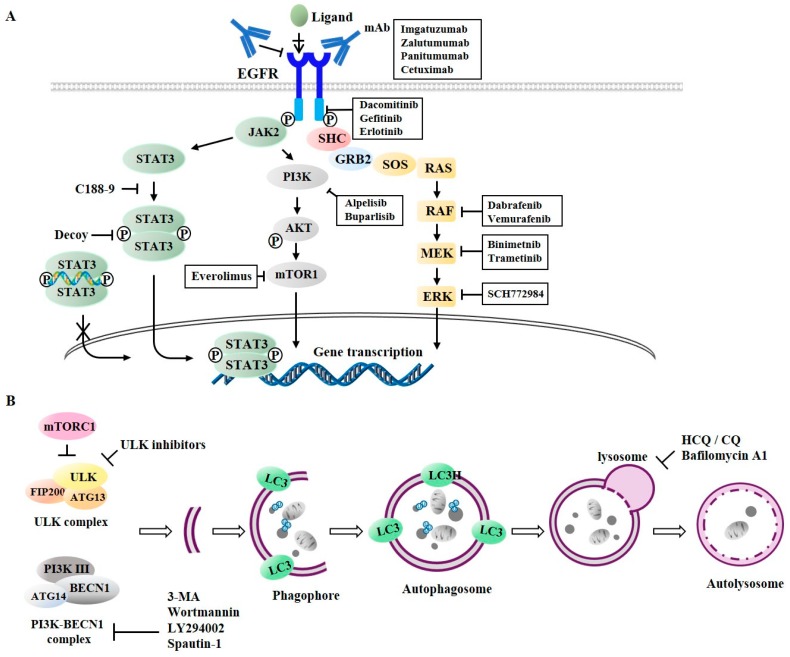
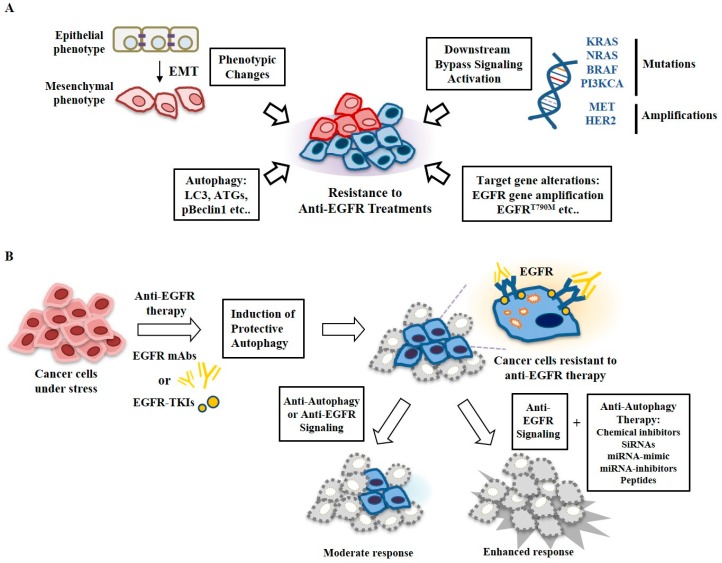
Epidermal growth factor receptor (EGFR) plays critical roles in cell proliferation, tumorigenesis, and anti-cancer drug resistance. Overexpression and somatic mutations of EGFR result in enhanced cancer cell survival. Therefore, EGFR can be a target for the development of anti-cancer therapy. Patients with cancers, including non-small cell lung cancers (NSCLC), have been shown to response to EGFR-tyrosine kinase inhibitors (EGFR-TKIs) and anti-EGFR antibodies. However, resistance to these anti-EGFR treatments has developed. Autophagy has emerged as a potential mechanism involved in the acquired resistance to anti-EGFR treatments. Anti-EGFR treatments can induce autophagy and result in resistance to anti-EGFR treatments. Autophagy is a programmed catabolic process stimulated by various stimuli. It promotes cellular survival under these stress conditions. Under normal conditions, EGFR-activated phosphoinositide 3-kinase (PI3K)/AKT serine/threonine kinase (AKT)/mammalian target of rapamycin (mTOR) signaling inhibits autophagy while EGFR/rat sarcoma viral oncogene homolog (RAS)/mitogen-activated protein kinase kinase (MEK)/mitogen-activated protein kinase (MAPK) signaling promotes autophagy. Thus, targeting autophagy may overcome resistance to anti-EGFR treatments. Inhibitors targeting autophagy and EGFR signaling have been under development. In this review, we discuss crosstalk between EGFR signaling and autophagy. We also assess whether autophagy inhibition, along with anti-EGFR treatments, might represent a promising approach to overcome resistance to anti-EGFR treatments in various cancers. In addition, we discuss new developments concerning anti-autophagy therapeutics for overcoming resistance to anti-EGFR treatments in various cancers.
Keywords: EGFR signaling; anti-EGFR treatments; autophagy; co-targeting.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Effects of hyperinsulinemia on acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitor via the PI3K/AKT pathway in non-small cell lung cancer cells in vitro.Oncol Lett. 2020 Nov;20(5):206. doi: 10.3892/ol.2020.12069. Epub 2020 Sep 8. Oncol Lett. 2020. PMID: 32963612 Free PMC article.
-
Preclinical rationale for PI3K/Akt/mTOR pathway inhibitors as therapy for epidermal growth factor receptor inhibitor-resistant non-small-cell lung cancer.Clin Lung Cancer. 2013 Jul;14(4):322-32. doi: 10.1016/j.cllc.2012.12.001. Epub 2013 Jan 16. Clin Lung Cancer. 2013. PMID: 23332287 Review.
-
Mechanisms of resistance to EGFR tyrosine kinase inhibitors.Acta Pharm Sin B. 2015 Sep;5(5):390-401. doi: 10.1016/j.apsb.2015.07.001. Epub 2015 Jul 26. Acta Pharm Sin B. 2015. PMID: 26579470 Free PMC article. Review.
-
Cotargeting EGFR and autophagy signaling: A novel therapeutic strategy for non-small-cell lung cancer.Mol Clin Oncol. 2014 Jan;2(1):8-12. doi: 10.3892/mco.2013.187. Epub 2013 Sep 18. Mol Clin Oncol. 2014. PMID: 24649300 Free PMC article. Review.
-
PPARgamma agonists sensitize PTEN-deficient resistant lung cancer cells to EGFR tyrosine kinase inhibitors by inducing autophagy.Eur J Pharmacol. 2018 Mar 15;823:19-26. doi: 10.1016/j.ejphar.2018.01.036. Epub 2018 Jan 31. Eur J Pharmacol. 2018. PMID: 29378193
Cited by
-
Fighting Drug Resistance through the Targeting of Drug-Tolerant Persister Cells.Cancers (Basel). 2021 Mar 5;13(5):1118. doi: 10.3390/cancers13051118. Cancers (Basel). 2021. PMID: 33807785 Free PMC article. Review.
-
Effect of EGFR on SQSTM1 Expression in Malignancy and Tumor Progression of Oral Squamous Cell Carcinoma.Int J Mol Sci. 2021 Nov 12;22(22):12226. doi: 10.3390/ijms222212226. Int J Mol Sci. 2021. PMID: 34830108 Free PMC article.
-
Combination of betulinic acid and EGFR-TKIs exerts synergistic anti-tumor effects against wild-type EGFR NSCLC by inducing autophagy-related cell death via EGFR signaling pathway.Respir Res. 2024 May 20;25(1):215. doi: 10.1186/s12931-024-02844-9. Respir Res. 2024. PMID: 38764025 Free PMC article.
-
ARID1A serves as a receivable biomarker for the resistance to EGFR-TKIs in non-small cell lung cancer.Mol Med. 2021 Oct 29;27(1):138. doi: 10.1186/s10020-021-00400-5. Mol Med. 2021. PMID: 34715776 Free PMC article. Review.
-
Radiogenomics of gastroenterological cancer: The dawn of personalized medicine with artificial intelligence-based image analysis.Ann Gastroenterol Surg. 2021 Feb 1;5(4):427-435. doi: 10.1002/ags3.12437. eCollection 2021 Jul. Ann Gastroenterol Surg. 2021. PMID: 34337291 Free PMC article. Review.
References
-
- Yao S., Shi F., Wang Y., Sun X., Sun W., Zhang Y., Liu X., Liu X., Su L. Angio-associated migratory cell protein interacts with epidermal growth factor receptor and enhances proliferation and drug resistance in human non-small cell lung cancer cells. Cell Signal. 2019;61:10–19. doi: 10.1016/j.cellsig.2019.05.004. - DOI - PubMed
-
- Wang H., Yao F., Luo S., Ma K., Liu M., Bai L., Chen S., Song C., Wang T., Du Q., et al. A mutual activation loop between the Ca(2+)-activated chloride channel TMEM16A and EGFR/STAT3 signaling promotes breast cancer tumorigenesis. Cancer Lett. 2019;455:48–59. doi: 10.1016/j.canlet.2019.04.027. - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
