

Degeneration of Injured Axons and Dendrites Requires Restraint of a Protective JNK Signaling Pathway by the Transmembrane Protein Raw
- PMID: 31492772
- PMCID: PMC6807270
- DOI: 10.1523/JNEUROSCI.0016-19.2019
Degeneration of Injured Axons and Dendrites Requires Restraint of a Protective JNK Signaling Pathway by the Transmembrane Protein Raw
Abstract
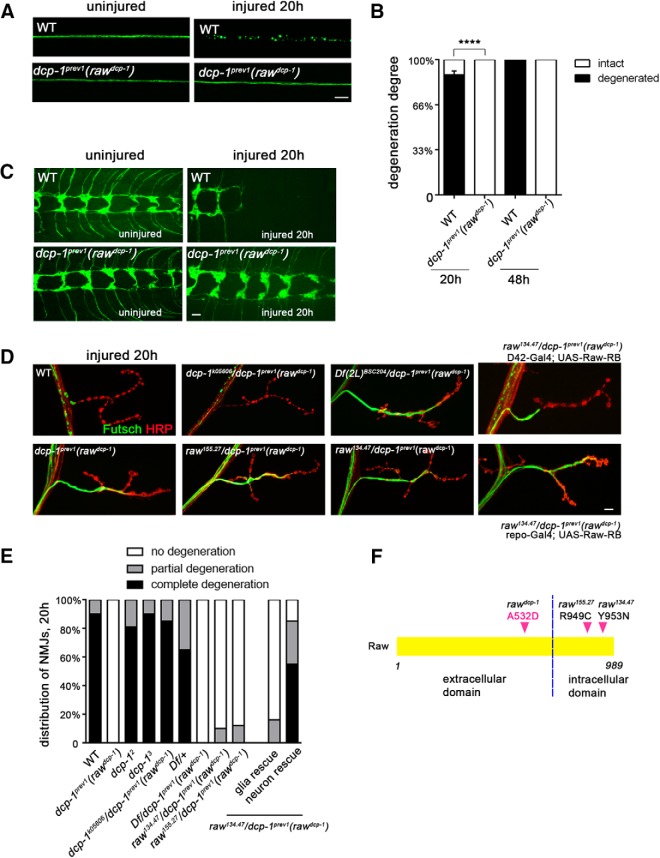
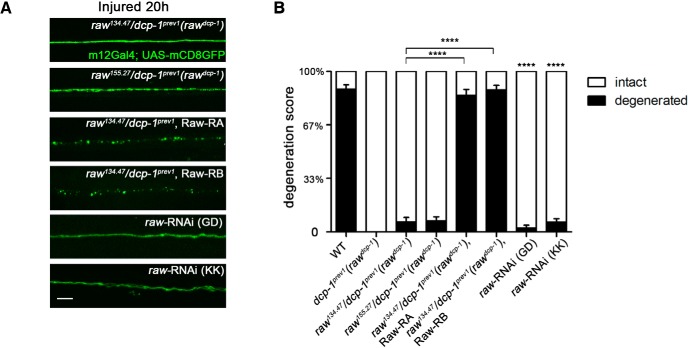
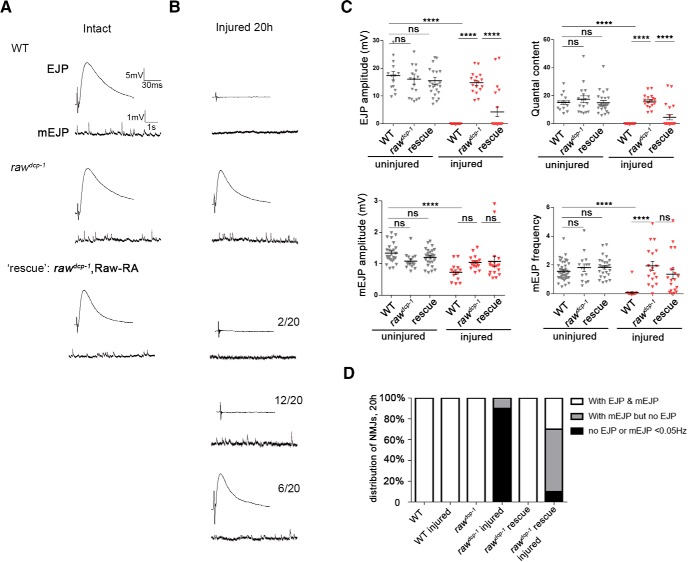
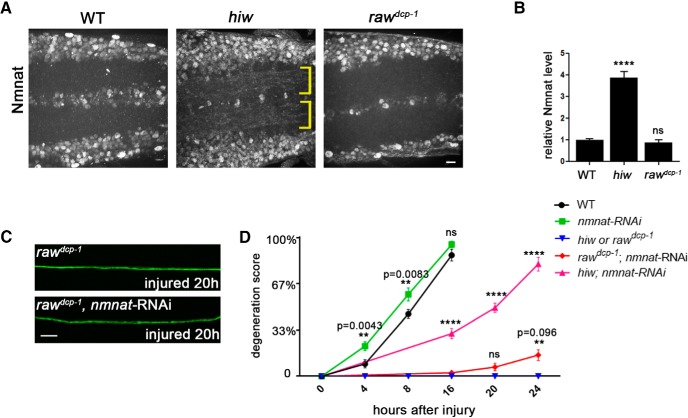
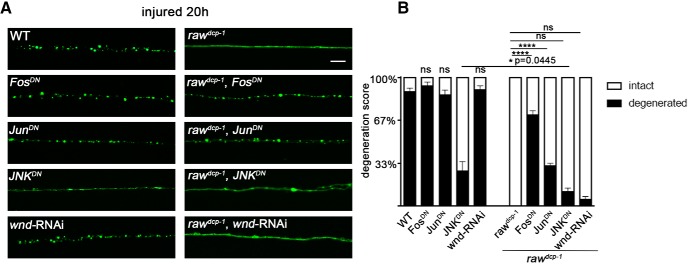
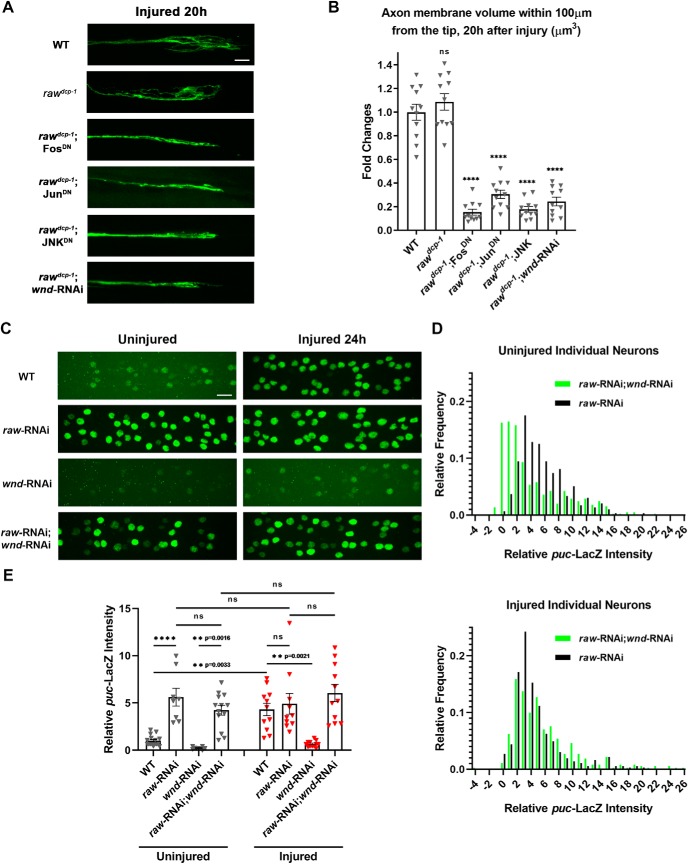
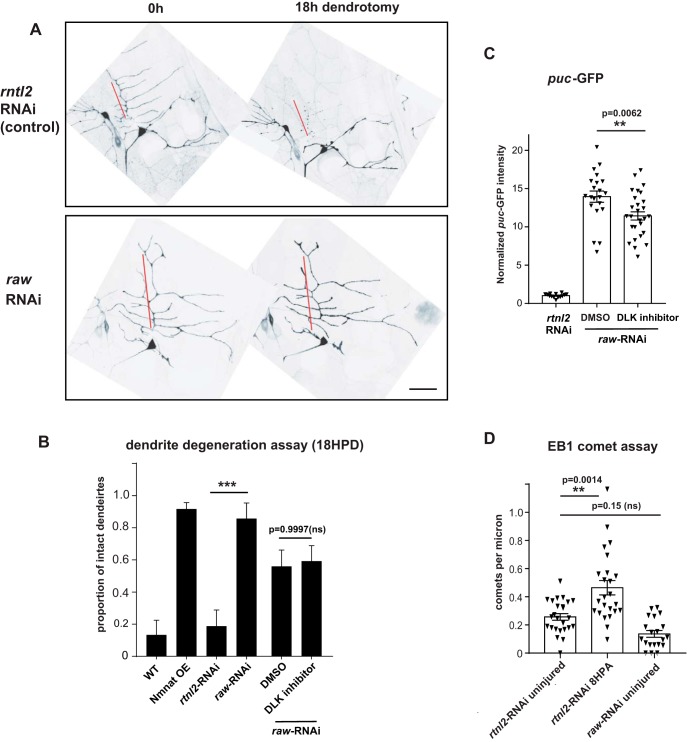
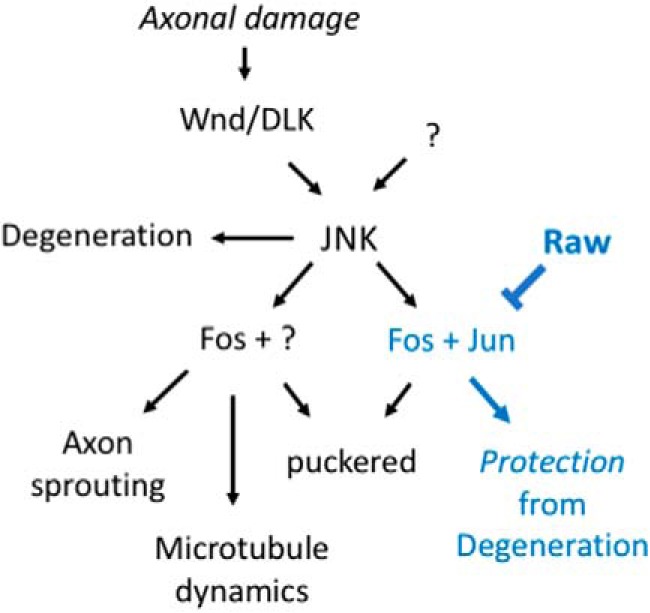
The degeneration of injured axons involves a self-destruction pathway whose components and mechanism are not fully understood. Here, we report a new regulator of axonal resilience. The transmembrane protein Raw is cell autonomously required for the degeneration of injured axons, dendrites, and synapses in Drosophila melanogaster In both male and female raw hypomorphic mutant or knock-down larvae, the degeneration of injured axons, dendrites, and synapses from motoneurons and sensory neurons is strongly inhibited. This protection is insensitive to reduction in the levels of the NAD+ synthesis enzyme Nmnat (nicotinamide mononucleotide adenylyl transferase), but requires the c-Jun N-terminal kinase (JNK) mitogen-activated protein (MAP) kinase and the transcription factors Fos and Jun (AP-1). Although these factors were previously known to function in axonal injury signaling and regeneration, Raw's function can be genetically separated from other axonal injury responses: Raw does not modulate JNK-dependent axonal injury signaling and regenerative responses, but instead restrains a protective pathway that inhibits the degeneration of axons, dendrites, and synapses. Although protection in raw mutants requires JNK, Fos, and Jun, JNK also promotes axonal degeneration. These findings suggest the existence of multiple independent pathways that share modulation by JNK, Fos, and Jun that influence how axons respond to stress and injury.SIGNIFICANCE STATEMENT Axonal degeneration is a major feature of neuropathies and nerve injuries and occurs via a cell autonomous self-destruction pathway whose mechanism is poorly understood. This study reports the identification of a new regulator of axonal degeneration: the transmembrane protein Raw. Raw regulates a cell autonomous nuclear signaling pathway whose yet unknown downstream effectors protect injured axons, dendrites, and synapses from degenerating. These findings imply that the susceptibility of axons to degeneration is strongly regulated in neurons. Future understanding of the cellular pathway regulated by Raw, which engages the c-Jun N-terminal kinase (JNK) mitogen-activated protein (MAP) kinase and Fos and Jun transcription factors, may suggest new strategies to increase the resiliency of axons in debilitating neuropathies.
Keywords: Drosophila; MAP kinase signaling; Wallerian degeneration; axon degeneration; axon injury; motoneuron.
Copyright © 2019 the authors.
Figures
Similar articles
-
SkpA restrains synaptic terminal growth during development and promotes axonal degeneration following injury.J Neurosci. 2014 Jun 18;34(25):8398-410. doi: 10.1523/JNEUROSCI.4715-13.2014. J Neurosci. 2014. PMID: 24948796 Free PMC article.
-
The Highwire ubiquitin ligase promotes axonal degeneration by tuning levels of Nmnat protein.PLoS Biol. 2012;10(12):e1001440. doi: 10.1371/journal.pbio.1001440. Epub 2012 Dec 4. PLoS Biol. 2012. PMID: 23226106 Free PMC article.
-
A conditioning lesion protects axons from degeneration via the Wallenda/DLK MAP kinase signaling cascade.J Neurosci. 2012 Jan 11;32(2):610-5. doi: 10.1523/JNEUROSCI.3586-11.2012. J Neurosci. 2012. PMID: 22238096 Free PMC article.
-
Why is NMNAT Protective against Neuronal Cell Death and Axon Degeneration, but Inhibitory of Axon Regeneration?Cells. 2019 Mar 21;8(3):267. doi: 10.3390/cells8030267. Cells. 2019. PMID: 30901919 Free PMC article. Review.
-
Models of axon regeneration in Drosophila.Exp Neurol. 2017 Jan;287(Pt 3):310-317. doi: 10.1016/j.expneurol.2016.03.014. Epub 2016 Mar 17. Exp Neurol. 2017. PMID: 26996133 Free PMC article. Review.
Cited by
-
JNK Signaling Pathway Involvement in Spinal Cord Neuron Development and Death.Cells. 2019 Dec 5;8(12):1576. doi: 10.3390/cells8121576. Cells. 2019. PMID: 31817379 Free PMC article. Review.
-
Opposing roles of Fos, Raw, and SARM1 in the regulation of axonal degeneration and synaptic structure.Front Cell Neurosci. 2023 Nov 30;17:1283995. doi: 10.3389/fncel.2023.1283995. eCollection 2023. Front Cell Neurosci. 2023. PMID: 38099151 Free PMC article.
-
Bidirectional Regulation of Sleep and Synapse Pruning after Neural Injury.Curr Biol. 2020 Mar 23;30(6):1063-1076.e3. doi: 10.1016/j.cub.2019.12.065. Epub 2020 Mar 5. Curr Biol. 2020. PMID: 32142703 Free PMC article.
-
HTT (huntingtin) and RAB7 co-migrate retrogradely on a signaling LAMP1-containing late endosome during axonal injury.Autophagy. 2023 Apr;19(4):1199-1220. doi: 10.1080/15548627.2022.2119351. Epub 2022 Sep 9. Autophagy. 2023. PMID: 36048753 Free PMC article.
-
A nerve-wracking buzz: lessons from Drosophila models of peripheral neuropathy and axon degeneration.Front Aging Neurosci. 2023 Aug 8;15:1166146. doi: 10.3389/fnagi.2023.1166146. eCollection 2023. Front Aging Neurosci. 2023. PMID: 37614471 Free PMC article. Review.
References
-
- Bauer Huang SL, Saheki Y, VanHoven MK, Torayama I, Ishihara T, Katsura I, van der Linden A, Sengupta P, Bargmann CI (2007) Left-right olfactory asymmetry results from antagonistic functions of voltage-activated calcium channels and the raw repeat protein OLRN-1 in C. elegans. Neural Dev 2:24. 10.1186/1749-8104-2-24 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous